Симптомы
Признаки болезни различны, они зависят от того, где именно локализованы амилоидные отложения, насколько сильно распространена болезнь, есть ли осложнения. Зачастую наблюдается комплекс симптомов, отражающих поражение нескольких органов.
При амилоидозе ЖКТ наблюдаются:
- увеличение языка;
- трудности с глотанием;
- нарушение стула;
- изжога, тошнота;
- боли в животе.
Признаки амилоидоза печени:
- изменение размеров печени;
- боли в подреберье справа;
- тошнота, отрыжка;
- желтуха.
Для амилоидоза поджелудочной присуща боль в левом подреберье слева тупого характера.
Амилоидоз сердца выражается в нарушении ритма, поражениях миокарда, сердечной недостаточности.
Амилоидоз нервной системы имеет следующие симптомы:
- периферическая полиневропатия (онемение конечностей, чувство покалывания, жжения);
- головные боли, головокружение, повышенное потоотделение;
- недержание мочи и кала;
- половая дисфункция.
При амилоидозе органов дыхания наблюдается хриплость голоса, бронхит.
Классификация и эпидемиология
При АС происходит инфильтрация миокарда нерастворимым гликопротеидом – амилоидом, образующимся из различных сывороточных или локально продуцируемых белковпредшественников. В их число входят константные белки, не зависящие от типа амилоида, например сывороточный амилоидный Р-белок (SAP), однако около 80 % массы амилоида составляют белки, отличающиеся при разных типах амилоида. Современная классификация амилоидоза основана на различиях амилоидогенных белков-предшественников. Таких белков в настоящее время известно около 30; соответственно, выделяют около 30 форм амилоидоза. Каждую форму обозначают аббревиатурой, включающей обозначение белка-предшественника: AL (L – легкие цепи иммуноглобулинов), ATTR (TTR – транстиретин), СAA (сывороточный амилоид А) и др. (см. таблицу). Согласно этой классификации, первичный и ассоциированный с миеломой амилоидоз следует обозначать аббревиатурой AL, т. к. амилоид при этих формах образован легкими цепями иммуноглобулинов. При системных формах амилоидоза (большинство случаев AL, а также ATTR, AA и др.) амилоидогенный белок-предшественник циркулирует в крови и может откладываться в различных тканях, тропность к которым отличается у разных белков. Кроме того, возможно развитие локального амилоидоза, например предсердий, вследствие местной продукции амилоидогенного варианта предсердного натрийуретического фактора (AANF).
. Классификация амилоидоза сердца.
Самым частым вариантом системного амилоидоза считают AL-амилоидоз – одну из многочисленных форм плазмоклеточной дискразии. Термин “плазмоклеточная дискразия” объединяет группу состояний, обусловленных патологией плазматических клеток костного мозга, реже – других тканей. При этом в костном мозге отмечается доминирование патологического клона плазматических клеток, синтезирующих аномальные иммуноглобулины или их компоненты: тяжелые или легкие цепи. Кроме AL-амилоидоза к плазмоклеточным дискразиям относят множественную миелому, макроглобулинемию Вальденстрема и др.
При AL-амилоидозе патологический клон плазматических клеток синтезирует легкие цепи иммуноглобулинов, осаждение которых в тканях сопровождается образованием нерастворимых фибрилл амилоида. AL-амилоидоз – наиболее тяжелая, генерализованная форма заболевания, поражающая многие ткани. Сердце поражается у 90 % больных, у половины из которых развивается диастолическая сердечная недостаточность (СН) [2]. Смерть обычно наступает от СН или аритмий [4].
Изолированный кардиопатический вариант AL-амилоидоза диагностируют редко – менее чем у 5 % больных [5]. Изолированное поражение сердца характерно для AANF-амилоидоза, при котором отмечают инфильтрацию предсердий амилоидом, образующимся из предсердного натрийуретического пептида. Эту форму амилоидоза обнаруживают более чем у 95 % людей старше 80 лет [6]. AANF-амилоидоз редко приводит к СН [2], но может стать причиной развития наджелудочковых аритмий [7–9].
Поражение сердца характерно для приобретенного и различных вариантов наследственного ATTR- амилоидоза. Эта форма обусловлена отложением белка-переносчика гормонов щитовидной железы и ретинола – транстиретина. Он секретируется печенью в тетрамерной форме, состоящей из четырех молекул транстиретина. С возрастом снижение активности ферментных систем гепатоцита приводит к повышению секреции амилоидогенных мономерных форм транстиретина и вследствие этого – раз- витию амилоидоза. Приобретенный ATTR-амилоидоз обычно называют системным старческим амилоидозом (поражает до 25–36 % людей старше 80 лет). Амилоид находят во многих органах, однако наиболее значимые депозиты обнаруживают в сердце. Обычно заболевание имеет доброкачественное малосимптомное течение, проявляется блокадой передней ветви левой ножки пучка Гиса и небольшим утолщением стенок левого желудочка (ЛЖ). При значительном отложении амилоида могут развиваться кардиомегалия и медленнопрогрессирующая СН [3].
При наличии мутаций в молекуле транстиретина сборка тетрамеров в гепатоците нарушена исходно, что приводит к развитию наследственного ATTR-амилоидоза в более раннем возрасте. В настоящий момент известно около 100 точечных мутаций и делеций гена транстиретина, 87 из которых амилоидогенны [10]. Для заболевания характерно аутосомно-доминантное наследование, однако пенетрантность мутантного гена варьируется в широких пределах. Выделяют невропатическую, кардиопатическую и офтальмолептоменингеальную формы заболевания. Чаще всего развивается невропатическая форма, которая характеризуется прогрессирующим поражением периферической и автономной нервной системы. Преимущественное поражение того или иного органа во многом связано с характером мутации. Так, при замене изолейцина в позиции 122 на валин развивается старческое амилоидное поражение сердца без неврологических проявлений; эту мутацию обнаруживают преимущественно у афроамериканцев [11]. К мутациям, вызывающим серьезное поражение сердца, относят замену метионина на валин в положении 30 (португальский вариант семейного амилоидоза), замену серина на изолейцин в положении 84 и замену аланина на треонин в положении 60. Амилоидная кардиопатия при наследственном ATTR-амилоидозе протекает менее тяжело, чем при AL-амилоидозе, но все же приводит к выраженной СН. При одном из наиболее распространенных вариантов амилоидоза – вторичном АА-амилоидозе, клинически значимое поражение сердца обнаруживают редко – менее чем в 10 % случаев [13]. Хотя АА-амилоидоз имеет системный характер, а при морфологическом исследовании депозиты амилоида обнаруживают в различных тканях, при этой форме заболевания клиническое значение имеет в основном поражение почек, в меньшей степени – печени, селезенки и желудочнокишечного тракта.
Лечение и профилактика
В большинстве случаев лечение амилоидоза проводится в домашних условиях. При наличии осложнений больному может быть показана госпитализация.
Терапия амилоидоза включает в себя приём препаратов и соблюдение ряда рекомендаций врача. Но в тяжелых случаях проводится удаление селезёнки, может потребоваться трансплантация почек или печени.
Перечень лекарств зависит от локализации отложений, степени поражения организма, существующих осложнений. Так, при вторичном амилоидозе необходимо специфическое лечение первичного заболевания. Кроме того, назначаются препараты для устранения симптоматики.
Также больному нередко показана специальная диета (ограничение приёма белка и соли).
Специфической профилактической программы амилоидоза не существует, так как точные причины возникновения заболевания неизвестны.
Амилоидоз —это комплексное заболевание, требующее постоянного лечения. Пациент с амилоидозом должен регулярно наблюдаться у специалиста и проходить обследования с целью контроля состояния здоровья. Медицинский обладает современным диагностическим оборудованием, что позволяет поставить точный диагноз в кратчайшие сроки. А опытные высококвалифицированные специалисты центра назначат эффективное лечение и обеспечат надлежащий уход за пациентом.
Амилоидоз сердца (обзор современных данных)
Номенклатура
Правильная номенклатура использует букву «А» для амилоида, за которой следуют буква (буквы), указывающая на основной депонируемый белок. Например, амилоидоз легкой цепи представляет собой «AL» («A» для амилоида и «L» для легкой цепи). Транстиретин-амилоидоз — «ATTR» («A» для амилоида и «TTR» для транстиретина). Такие термины, как «первичный амилоидоз», «вторичный амилоидоз», «сенильный амилоидоз» и «семейная амилоидная кардиомиопатия», часто приводят к путанице и их, как правило, следует избегать.
Подавляющее большинство сердечного амилоидоза вызвано одним из двух белков: легкими цепями или транстиретином. Далее следует обсуждение презентаций и конкретных методов лечения для этих двух подтипов.
Light-Chain (AL) Амилоидоз
Наиболее часто диагностируемой формой системного амилоидоза является AL амилоидоз. Полное обсуждение патофизиологии заболевания выходит за рамки этого обзора, но краткое рабочее понимание полезно, чтобы лучше понять варианты лечения.
Плазматические клетки находятся в основном в костном мозге и вырабатывают большое количество антител. Антитела состоят из тяжелых цепей и легких цепей. Когда плазматическая клетка становится клональной (по существу становится злокачественной), она и ее клоны обычно продуцируют клональное антитело и клональный избыток легкой цепи, связанный с этим антителом. На данный момент, есть три возможных результата:
Клон плазматических клеток захватывает небольшую часть костного мозга, и легкая цепь безвредно выводится с мочой. Это состояние называется моноклональной гаммопатией неопределенного значения.
Клон плазматических клеток занимает большую часть костного мозга, что может привести к гиперкальциемии, анемии, литическим поражениям и / или почечной дисфункции. Это состояние называется миеломой.
Клон плазматических клеток производит легкую цепь, которая склонна к неправильному складыванию в бета-плиссированные листы. Эти легкие цепи циркулируют в кровотоке и откладываются в одной или нескольких тканях. Это состояние называется AL амилоидоз.
Отложения амилоида AL могут встречаться практически в любом органе, и характер поражения органов варьируется от пациента к пациенту. Распространенные участки вовлечения и связанные с ними проявления включают почки (альбуминурия и потенциальная почечная недостаточность), печень (повышение уровня щелочной фосфатазы и потенциальная печеночная недостаточность), желудочно-кишечный тракт (ЖКТ) (дисфагия, запор, мальабсорбция и желудочно-кишечное кровотечение), язык ( макроглоссия) и нервы (периферическая невропатия и вегетативная дисфункция). Пациенты редко имеют клиническое поражение всех этих систем органов.
Сердечные проявления включают сердечную недостаточность (СН) (диастолическая> систолическая) и аритмии (тахиаритмия / брадиаритмия). Ключом к диагнозу является желудочковая «гипертрофия», наблюдаемая на эхокардиографии с неадекватно мало выраженными электрическими изменениями на электрокардиограмме (ЭКГ). Натрийуретический пептид B-типа и натрийуретический пептид pro-B-типа N-конца обычно повышен при сердечном амилоидозе, и анализы тропонина часто являются хронически положительными при низких уровнях (0, 1-1 нг / мл) из-за продолжающегося постепенного разрушения кардиомиоцитов.
Транстиретин (АТТР) амилоидоз
Транстиретин (преальбумин) является обильным белком, вырабатываемым печенью, и функционирует в качестве переносчика тироксина и ретинола. Обычно он циркулирует преимущественно как гомотетрамер с небольшим количеством транстиретина, циркулирующего в мономерной форме. Мономерная форма транстиретина склонна к неправильному складыванию и постепенно откладывается в виде амилоидных отложений.
Существуют два основных подтипа амилоидоза ATTR: ATTR природного типа и мутантный ATTR. В случае ATTR природного типа белок транстиретина является нормальным (не мутированным) белком и в течение десятилетий постепенно откладывается в виде амилоидных отложений. Хотя небольшие количества отложений могут возникать в мягких тканях (вызывая синдром запястного канала) и сосудистой сети, первичные патологические отложения возникают в сердце.
Пациенты с мутантным ATTR были рождены с патологической мутацией в гене транстиретина, приводящей к ускоренному отложению амилоида. Мутантный ATTR чаще всего откладывается в сердце и / или нервах, причем характер отложения в значительной степени зависит от мутации. В Соединенных Штатах наиболее часто встречающейся мутацией является мутация V122I; он присутствует у 3-3, 5% лиц африканского происхождения. Точная пенетрантность мутации неизвестна и, вероятно, <10% 1, но это почти наверняка является сильно недиагностированной формой сердечной недостаточности в афро-американской популяции.
Диагностика
Окончательный диагноз амилоидоза требует биопсии. В качестве места иногда выбирают подкожную клетчатку живота из-за легкости и низкой травмоопастности процедуры. Тем не менее, этот метод страдает от относительно низкой чувствительности; даже в положительных случаях часто бывают неадекватные амилоидные отложения, чтобы окончательно типировать заболевание (ATTR, AL и т. д. ). Таким образом, общая практика в Стэнфордском амилоидном центре заключается в биопсии клинически вовлеченного органа (то есть сердца при подозрении на амилоидоз сердца), потому что эта практика имеет почти 100% чувствительность / специфичность и практически всегда допускает окончательный подтип. Подтипирование может быть выполнено с помощью иммунофлуоресценции или отправлено в реферальную лабораторию для масс-спектрометрии. Если есть какие-либо сомнения относительно диагноза, основанного на иммунофлуоресцентном окрашивании, следует выполнить масс-спектрометрию.
Лабораторные анализы на наличие амилоидоза AL включают анализ дисфункции других органов (например, протеинурии, щелочной фосфатазы) и непосредственное измерение циркулирующих легких цепей. Анализ свободных легких цепей может быть полезен как для оценки вероятности диагноза до биопсии (например, нормальные циркулирующие легкие цепи делают диагноз амилоидоза AL маловероятным), так и для мониторинга реакции заболевания на химиотерапию.
При амилоидозе ATTR важно предложить генетическое тестирование гена транстиретина. Наличие патологической мутации может повлиять на варианты клинических исследований, предсказать места поражения органов и иметь отношение к членам семьи.
При любой форме сердечного амилоидоза доминирующим признаком визуализации является появление «гипертрофии» сердца. «Гипертрофия» при визуализации представляет собой отложение амилоидных фибрилл, а не гипертрофию / гиперплазию миоцитов, что объясняет, почему изменения ЭКГ не выражены. Важно отметить, что степень гипертрофии является переменной, поэтому у пациента без классических результатов визуализации важно поддерживать клиническое подозрение в соответствующих обстоятельствах (например, необъяснимая сердечная недосоаточность у пациента с миеломой или необъяснимая сердечаня недостаточность у пациента с протеинурией / макроглоссия).
Сердечный амилоидоз вызывает аномальные паттерны позднего усиления гадолиния по сердечному магнитному резонансу (CMR) как в глобальном трансмуральном, так и в субэндокардиальном распределении. Часто встречаются также подъемы во время спин-решеточной релаксации миокарда на CMR. Трансторакальная эхокардиография обычно выявляет базальные> преобладающие апикальные нарушения при их деформации. Хотя степень CMR и аномалии деформации коррелировали с исходами у пациентов, на сегодняшний день трудно с уверенностью сказать, что они добавляют дополнительную информацию по сравнению с другими прогностическими моделями. Кроме того, эти модели все же не достаточно специфичны для диагностики амилоидоза, чтобы избежать необходимости постановки точного диагноза, подтвержденного биопсией.
Роль повторных оценок позднего усиления гадолиния или времен спин-решеточной релаксации на CMR или оценки деформации с использованием эхокардиографии неясна. Что касается амилоидоза AL, неясно, что данный мониторинг значительно улучшает оценку реакции на заболевание, используя изменения в легких сывороточных цепях и биомаркерах сердца.
Прогноз
Прогноз при амилоидозе зависит, прежде всего, от степени поражения сердца и, при AL-амилоидозе, от уровня циркулирующих легких цепей. Поскольку возможности химиотерапии в последние годы значительно расширились, прогноз для амилоидоза AL также заметно улучшился, при этом ожидаемая продолжительность жизни большинства пациентов, включая многих из них со значительным поражением сердца, измеряется годами, а не месяцами. Прогноз в ATTR амилоидоз, как правило, лучше, чем при амилоидозе AL, хотя обе формы заболевания все еще имеют высокую годовую смертность.
Для AL амилоидоза были предложены различные системы стадирования, большинство из которых сосредоточено в основном на степени поражения сердца. Широко используемая система стадирования, опубликованная в 2004 году, основывалась исключительно на двух биомаркерах сердца: тропонине (T или I) и N-терминальном натрийуретическом пептиде про-B-типа. Степень оценки была оценена как 1, 2 или 3 в зависимости от того, 0, 1 или 2 биомаркера были повышены. Этапная система успешно выявила четкие кривые выживаемости, хотя общая выживаемость оставалась неутешительной во всех 3 группах с медианной выживаемостью 26, 4, 10, 5 и 3, 5 мес на стадиях 1, 2 и 3 соответственно. Пересмотренная система этапов была опубликована той же группой в 2012 году, добавив абсолютную разницу в свободных легких цепях в качестве третьей переменной, таким образом, разделив пациентов на 4 этапа. В дополнение к повышению точности этапов, эта система продемонстрировала улучшения в целом. Выживаемость на всех стадиях со средней выживаемостью 94, 1, 40, 3, 14 и 5, 8 месяцев для стадий 1, 2, 3 и 4 соответственно. С продолжающимся развитием химиотерапии, вероятно, что выживание данных пациентов продолжит существенно улучшаться.
Лечение
Лечение АЛ-амилоидоза происходит по двум параллельным путям: лечение последствий дисфункции органов при одновременной попытке замедлить прогрессирование заболевания путем уничтожения клональных плазматических клеток (и, следовательно, сокращения циркулирующих патологических легких цепей) с помощью химиотерапии.
Специфическое для сердца лечение всех форм амилоидоза в значительной степени включает управление объемом (диуретики / ограничение соли) и управление аритмией. Нейрогормональные антагонисты, обычно используемые при СН, часто плохо переносятся и контрпродуктивны. В частности, бета-блокаторы, ингибиторы ангиотензин-превращающего фермента и блокаторы рецепторов ангиотензина часто приводят к гипотонии (из-за вегетативной дисфункции и наличия небольшой полости левого желудочка с неспособностью увеличивать ударный объем в ответ на вазодилатацию), и бета-блокаторы часто усугубляют брадиаритмию. Дигоксин связывается с амилоидными фибриллами, что приводит к потенциальной токсичности дигоксина даже при нормальных уровнях в крови, и его, как правило, следует избегать.
Кардиостимуляторы часто необходимы из-за высокой распространенности заболеваний, связанных с проводимостью (особенно при амилоидозе ATTR). Мерцательная аритмия встречается часто и часто плохо переносится, часто требуя кардиоверсии и / или антиаритмической терапии (чаще всего амиодарона). Желудочковые аритмии и внезапная сердечная смерть также распространены. Исторически сложилось так, что имплантируемые кардиовертер-дефибрилляторы (ICD) не поощрялись из-за плохого прогноза, связанного с амилоидозом сердца, но более поздние данные свидетельствуют о том, что ICD могут быть эффективной частью стратегии лечения у пациентов с разумным прогнозом от амилоидоза сердца (> 1 года). )
Поскольку сердечный амилоидоз необратим и часто может быть связан с тяжелыми симптомами и высокими показателями смертности, сердечно-сосудистую трансплантацию следует рассматривать у хорошо отобранных пациентов. Трансплантация сердца должна сопровождаться стратегией на основе химиотерапии для контроля производства легких цепей. При амилоидозе ATTR важно исключить значительную неврологическую вовлеченность пациентов с семейными формами. Во всех случаях мы рекомендуем проводить трансплантацию только в центрах, где есть опыт трансплантации пациентов с амилоидозом.
За последнее десятилетие варианты химиотерапии при амилоидозе AL заметно улучшились, так что большинство пациентов могут достичь значительного снижения (и иногда полной нормализации) своих циркулирующих патологических легких цепей. Химиотерапия обычно включает комбинацию нескольких классов противоопухолевых средств, включая алкилаторы (например, мелфалан или циклофосфамид), стероиды (например, дексаметазон), ингибиторы протеаз (например, бортезомиб или карфилзомиб) и / или иммуномодуляторы (например, леналидомид). Альтернативная стратегия включает пересадку ауто-стволовых клеток с использованием высокодозной химиотерапии с алкилирующим агентом. Хотя трансплантация ауто-стволовых клеток может быть эффективным методом снижения легких цепей и используется во многих центрах, единственное рандомизированное исследование, сравнивающее его со стандартной химиотерапией, показало, что трансплантация ауто-стволовых клеток имеет худшие результаты. Стандартные варианты химиотерапии в течение последнего десятилетия, практика нашего центра заключается в том, чтобы проводить трансплантацию ауто-стволовых клеток лишь в редких случаях.
Поскольку ATTR амилоидоз не является злокачественным процессом, химиотерапия не играет никакой роли. Хотя в настоящее время нет утвержденных препаратов, модифицирующих заболевание, для лечения амилоидоза АТТР, несколько препаратов были изучены и / или находятся на поздней стадии испытаний. К ним относятся следующие:
Дифлунизал. Этот нестероидный противовоспалительный препарат, одобренный для лечения боли при артрите, стабилизирует тетрамерную форму транстиретина. Рандомизированное исследование продемонстрировало замедление прогрессирования заболевания среди пациентов с полинейропатией из-за мутантного амилоидоза ATTR. Поскольку нестероидные противовоспалительные препараты сравнительно противопоказаны при СН, это вряд ли будет хорошим вариантом для кардиомиопатии ATTR.
Tafamidis. Этот агент одобрен в некоторых частях мира (Европа и Япония) для мутантного амилоидоза ATTR, вызывающего полинейропатию, но не одобрен в Соединенных Штатах. Фаза 3 исследования, посвященная изучению его полезности при кардиомиопатии ATTR (как ATTR природного типа, так и мутантного ATTR), завершила регистрацию, и результаты ожидаются.
РНК-интерференция. Два агента, которые действуют через небольшие интерферирующие РНК или РНК-интерференции (уменьшая выработку транстиретина печенью), находятся в фазе 3 испытаний на амилоидоз ATTR, включая как полиневропатию, так и кардиомиопатические формы.
Резюме
Парадигма сердечного амилоидоза заметно изменилась за последнее десятилетие, все большее количество пациентов, диагностируемых в плане амилоидоза и с заметными улучшениями в терапевтических вариантах. Диагностические признаки включают необъяснимую гипертрофию желудочков с невыражеными изменениями ЭКГ и необъяснимую СН, возникающую с характерной дисфункцией других органов. Окончательный диагноз требует биопсии и имеет решающее значение для окончательного подтипирования амилоидных отложений (например, AL и ATTR).
В случае амилоидоза AL лечение должно начинаться незамедлительно с химиотерапии (наш предпочтительный вариант) или аутотрансплантации стволовых клеток. В случае амилоидоза ATTR пациенты должны быть направлены для оценки для включения в одно из текущих клинических испытаний. Кардиологическая коррекция в первую очередь включает диуретики / ритм-контроль. Для отдельных пациентов следует рассмотреть возможность имплантации ICD или трансплантации сердца.
Поскольку заболевание, как правило, требует многопрофильной экспертизы, мы рекомендуем, когда это возможно, обращаться в амилоидный центр для консультации.
Лечение
Фото: sodrugestvo.info
Целью лечения амилоидоза вне зависимости от типа и особенностей течения заболевания считается снижение концентрации особых белков-предшественников, которые в последующем превращаются в амилоид и откладываются в тканях. Кроме того важнейшей задачей является стабилизация или восстановление функций внутренних органов, предупреждение развития органной недостаточности и повышение продолжительности жизни пациентов.
Симптоматические мероприятия при амилоидозе направлены на устранение уже возникших нарушений: отеков, аритмии, гипотензии, сердечной недостаточности и пр. Лечение амилоидоза, в основном, консервативное. При развитии осложнений, критическом снижении функций жизненно важных органов осуществляются хирургические вмешательства.
Схемы лечения варьируются в зависимости от типа болезни:
- AA-амилоидоз. Рекомендована противовоспалительная терапия в сочетании с оперативными методиками при развитии локальных гнойных процессов.
- AL-амилоидоз. Необходимо подавление клона плазматических клеток, секретирующих особые субъединицы иммуноглобулинов (легкие цепи). Прекращение синтеза таких соединений и их накопления в тканях способствует выведению излишков амилоида, остановке прогрессирования патологии.
- ATTR-амилоидоз. Долгое время единственным эффективным методом лечения данной разновидности заболевания считалась пересадка печени, выделяющей неизмененный транстиретин. В последние годы на ранних стадиях болезни с достаточным успехом применяется терапия препаратом из группы селективных стабилизаторов транстиретина.
Медикаментозная терапия
При вторичной форме болезни требуется патогенетическая терапия, направленная на устранение или облегчение течения основного заболевания. Перечень лекарственных препаратов, дозы и продолжительность курсов определяются индивидуально с учетом характера и тяжести причинной патологии. При АА-амилоидозе необходима длительная, иногда – пожизненная базисная терапия, которую при развитии осложнений дополняют антибактериальными препаратами.
Пациентам с AL-амилоидозом показана высокодозная химиотерапия. Достижение ремиссии оценивают на основании лабораторных показателей. С учетом имеющейся симптоматики при любых формах амилоидоза производят медикаментозное устранение нарушений работы сердца и сосудов, осуществляют коррекцию давления и кишечной моторики, проводят борьбу с болевым синдромом.
Хирургическое лечение
При жизнеугрожающих нарушениях сердечного ритма требуется установка водителя ритма. При развитии локальных гнойных процессов на фоне AA-амилоидоза показано вскрытие, дренирование гнойников в мягких тканях, легких, суставах. При ATTR-амилоидозе осуществляют трансплантацию печени. Методика особенно эффективна у людей младше 50 лет с отсутствием осложнений со стороны сердечно-сосудистой системы.
Поскольку при системном амилоидозе нередко поражаются почки с развитием почечной недостаточности, ряду больных требуется трансплантация данного органа. Операция рекомендована при отсутствии грубых изменений сердца и ЖКТ. Пятилетняя выживаемость после пересадки составляет около 65%, что сопоставимо с показателями пациентов с ХПН, развившейся вследствие других заболеваний. У трети реципиентов в пересаженной почке в последующем также развивается амилоидоз, но отторжение трансплантата в результате отложения амилоида происходит всего в 2-3% случаев.
Заместительная почечная терапия
Хроническая почечная недостаточность является одной из ведущих причин гибели пациентов с системным амилоидозом. С учетом этого обстоятельства, при серьезном нарушении функции почек высокую значимость приобретают методы экстракорпорального очищения крови. По мнению специалистов, предпочтительным вариантом является перитонеальный диализ, сопряженный с меньшим риском падения давления во время проведения процедуры. Продолжительность жизни при использовании гемодиализа и перитонеального диализа такая же, как у людей с другими заболеваниями почек. Хорошие или удовлетворительные клинические и лабораторные результаты заместительной почечной терапии отмечаются в 60% случаев.
Морфология и патогенез
Отложение амилоида приводит к значительному концентрическомуутолщению миокарда, причем для АС не характерна дилатация полостей желудочков. Амилоидная инфильтрация миокарда сопровождается ухудшением его механических свойств и приводит к значительному снижению сократимости в сочетании с тяжелыми рестриктивными нарушениями диастолической функции. Современные методы исследования показали, что утолщение стенок миокарда при АС не всегда коррелирует с нарушением систолической и диастолической функций. Условно можно выделить 3 подтипа АС – у больных AL, наследственным ATTR и приобретенным сенильным ATTR-амилоидозом [16].
Во всех случаях амилоид обнаруживают в интерстиции миокарда в виде диффузных или узловых депозитов. Наиболее часто выявляют диффузное утолщение межжелудочковой перегородки (> 80 % больных) и задней стенки ЛЖ. Изолированное утолщение стенки правого желудочка (без утолщения стенки ЛЖ) встречается редко – в 6 % случаев, а утолщение стенок обоих желудочков – в 40–80 % случаев [3, 18]. Утолщение межпредсердной перегородки отмечается у 40 % больных. Изменение формы ЛЖ происходит по типу концентрической гипертрофии, дилатация желудочков не развивается [19]. Сочетание утолщения стенок миокарда более чем у половины больных с низкой амплитудой желудочкового комплекса на электрокардиограмме (ЭКГ) в грудных отведениях (менее 10 мм) и отведениях от конечностей (менее 5 мм) позволяет подтвердить псевдогипертрофический характер изменений миокарда. По данным C. Rapezzi и соавт., толщина стенок миокарда у больных ATTR-амилоидозом (особенно старческим) была достоверно больше, чем у пациентов с AL-амилоидозом [16, 20]. В то же время у больных AL-амилоидозом отмечено более значительное снижение амплитуды желудочкового комплекса и, соответственно, более низкое отношение амплитуды QRS к толщине стенки ЛЖ. Это может указывать на связь снижения амплитуды QRS у больных AL-амилоидозом в бóльшей степени с повреждением кардиомиоцитов, чем с интерстициальной депозицией амилоида. Известно, что в патогенезе повреждения органов у больных AL-амилоидозом большое значение имеет прямое токсическое действие свободных легких цепей иммуноглобулинов на клетки [16, 18]. У больных с разными типами АС обнаруживают значительную вакуолизацию кардиомиоцитов. Исследования последних лет показали [24], что именно вакуолизацией кардиомиоцитов, а не депозитами амилоида обусловлена повышенная эхогенность зернистого типа, характерная для АС и впервые описанная А. Siqueira-Filho и соавт. [19]. Таким образом, это ультразвуковое свойство амилоидного сердца не является специфичным и не может быть использовано в качестве диагностического критерия.
Отражением повреждения кардиомиоцитов у больных АС может быть повышение концентрации тропонинов и натрийуретических пептидов. Было показано, что экспрессия предсердного и мозгового нитрийуретических пептидов повышена в кардиомиоцитах желудочков у пациентов с АС, особенно в участках, прилегающих к депозитам амилоида [25]. Хотя указанные субстанции не являются специфическими маркерами амилоидного поражения сердца, их считают высокочувствительными показателями и, следовательно, важным фактором прогноза при системном амилоидозе [26].
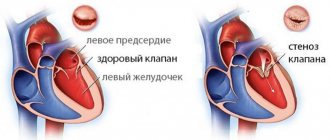
Амилоид может откладываться в области клапанов сердца, часто вызывая их утолщение, заметное на эхокардиограмме – Эхо-КГ [18, 27]. Описано утолщение папиллярных мышц [28], однако функция клапанов чаще сохранена.
Амилоид способен поражать проводящую систему сердца, описаны случаи проникновения амилоидных масс непосредственно в синоатриальный узел. Нарушения проводимости чаще всего представлены неполной блокадой передней ветви левой ножки пучка Гиса (20 %), полной блокадой правой (4–19 %) или левой ножки (2–7 %), атриовентрикулярной блокадой I степени (18–33 %) [16, 18]. Нарушения проводимости, как правило, встречаются при старческом АС, что, возможно, связано с более длительным течением данного варианта заболевания [3]. Отложение амилоида в области адренергических синапсов нарушает нейрогуморальную регуляцию работы сердца [29], что может быть одним из факторов риска тахиаритмий. У 5–27 % больных выявляют фибрилляцию предсердий, более редкими нарушениями ритма являются желудочковая тахикардия, узловой ритм. Какие-либо изменения на ЭКГ встречаются почти у всех (> 90 %) больных АС.
Отличительным свойством AL-амилоидоза является его отложение в стенках коронарных артерий [15], что может вызывать ишемию миокарда [30]. Некоторые больные жалуются на стенокардию [31], при этом изменения на ангиограмме могут отсутствовать [32]. Может развиваться инфаркт миокарда. Патологические Q-зубцы и изменения реполяризации без клинических признаков инфаркта миокарда или указания на него в анамнезе обнаруживают более чем у половины больных, что позволяет интерпретировать эти изменения как псевдоинфарктные. По-видимому, имитация инфарктных изменений связана с узловым отложением электрически неактивного амилоида, что создает эффект рубца. На поздних стадиях заболевания почти у половины больных выявляют перикардиальный выпот [18].
Количественная оценка совокупной массы амилоидных депозитов в сердце возможна с помощью SAP- сцинтиграфии. SAP – это нормальный гликопротеин плазмы, уровень которого резко возрастает в амилоидных депозитах любого типа в результате обратимого кальций-зависимого связывания с амилоидной фибриллой. После внутривенного введения радиофармпрепарат с меченым SAP рас- пределяется между циркулирующим и связанным с амилоидом пулами SAP пропорционально их объему. Таким образом, можно получить изображения для качественной и количественной оценки амилоидных депозитов. Метод особенно полезен при мониторировании течения амилоидоза, в т. ч. с целью оценки эффективности лечения.
Протекание заболевания и прогноз
Специалисты утверждают, что в основном причиной смертельного исхода больных является хроническая почечная недостаточность, которая наступает в результате реактивного амилоидоза, характеризующий как воспалительный инфекционный процесс. А также существуют сведения, что больные при вспышке реактивного амилоидоза умирают в результате инфекционного заболевания кишечника и диареи.
Статистические данные указывают, что даже при своевременном обнаружении и лечении заболевания средняя выживаемость пациентов с почечной недостаточностью составляет не более полтора года с момента обследования. На сегодняшний день ведущие специалисты продлевают жизнь больным посредством пересадки костного мозга, а также проводят хирургические процедуры по пересадке почки.
#диагностика #урология проблемы с почками
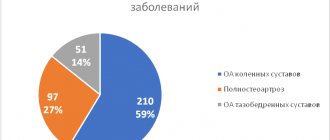
Классификация амилоидоза почек
Систематизация заболевания основывается на таких параметрах, как этиология и патогенез. Исходя из них, выделяют пять её форм.
| Форма | Характерные особенности |
| Врождённая | AL-амилоидоз, развитие которого было спровоцировано появлением и скоплением в тканях организма лёгких цепей иммуноглобулинов. Этиология заболевания до сих пор остаётся невыясненной, так же как и механизм его развития. |
| Приобретённая | АА-амилоидоз является вторичным, вызванным рядом хронических воспалительных процессов, при которых печень начинает усиленно производить белок острой фазы альфа-глобулина. |
| Семейная | AF-амилоидоз (или «средиземноморская лихорадка») является наследственной. Её болеют жители Средиземноморья (греки, арабы, армяне). В основе заболевания заложен генетический дефект, провоцирующий нарушение производства фибриллярных белков. |
| Старческая | ASC1-амилоидоз развивается у пациентов преклонного возраста из-за нарушения метаболизма сывороточного преальбумина. В соответствии с медицинской статистикой, заболевание поражает около 80% пациентов старше 80-ти лет. |
| Местная опухолевидная | AE-амилоидоз может быть спровоцирован новообразованиями эндокринной системы, сахарным диабетом второго типа, сенильной деменцией альцгеймеровского типа. |
Диагностика
Фото: pluska.sk
Современные методы диагностики амилоидоза
Амилоидоз – это заболевание, проявляющееся как патология белкового обмена и работоспособности иммунной системы. Амилоид (белково- сахаридный комплекс), появляющийся в результате этого нарушения, способен откладываться в клетках любых тканей организма. С развитием заболевания он постепенно заменяет здоровые клетки, и орган перестает функционировать. При тяжелой форме патологии наступает полиорганная недостаточность (поражение 50% и больше органов), приводящая к смерти.
Наследственная форма заболевания наблюдается в ареале Средиземного моря, а также у людей еврейской и армянской национальности. Среди мужчин данное заболевание встречается в два раза чаще.
Из наиболее распространенных форм заболевания можно назвать нефропатическую, при которой отложения наблюдаются в почках, и системную – отложения амилоида встречаются во многих органах.
Амилоид, откладывающийся в различных органах, отличается по структуре. Всего существует около 15 типов, различающихся по структуре и составу. Их основу составляют два типа:
- AA-амилоид. Хронические воспалительные заболевания вызывают высокий уровень содержания в плазме белка SAA и поддерживают его длительный период. При неполном расщеплении белка формируется фибриллярный AA-амилоид.
- AL-амилоид. Белки данного типа появляются при расщеплении амилоидобластов (переродившиеся плазмоциты). Они являются аномальными соединениями иммуноглобулина.
- Появление прочих типов амилоида определяется формой амилоидоза.
Виды заболевания
Амилоидоз встречается как самостоятельное, так и сопутствующее заболевание. Диагностика амилоидоза выделяет несколько видов:
Первичный или идиопатический. При подобном виде отложения встречаются во всех внутренних органах. Определить точную причину появления невозможно. Наблюдаются множественные аномалии в клетках иммунной системы с накоплением AL- амилоида в коже, в тканях мышечной, нервной и сердечно-сосудистой систем. Его причиной может стать плазмоцитома (миеломная болезнь) – злокачественная опухолевая патология.
Симптомы первичного амилоидоза:
- Миастения с последующей атрофией мышц;
- Диспепсия и диарея;
- Патология мочеполовой и репродуктивной системы;
- Поражение органов зрения.
Вторичный
Является осложнением какого-либо воспалительного заболевания. Причиной его появления могут стать:
- Хронические инфекционные заболевания: пиелонефрит, туберкулез, бронхоэктатическая болезнь, малярия, сифилис или проказа (лепра);
- Хронические гнойные заболевания: остеомиелит, гнойные язвы и раны;
- Язвенный колит – воспаление толстой кишки;
- Опухолевые поражения органов кроветворения: лейкоз, лимфогранулематоз и т.д.;
- Ревматологическая патология: различные артриты и прочее.
Вторичный амилоидоз образуется во внутренних органах. Наблюдается нарушение деятельности органов с наибольшим отложением амилоида – в области почек, селезенки, печени или лимфоузлов. В дальнейшем поражение распространяется на остальные органы с последующим смертельным исходом.
Наследственный
Подобная форма образуется из-за генетических аномалий в клетках иммунной системы, приводящих к появлению амилоидобластов. Подобная патология диагностируется в некоторых национальных группах или в определенной географической области. К наследственной форме можно отнести:
- Периодическая болезнь – семейная лихорадка Средиземноморья;
- Семейный нефропатический или английский амилоидоз;
- Наследственный нейропатический амилоидоз – португальский, американский или финский;
- Наследственный кардиопатический или датский амилоидоз.
Старческий
Системный подход позволяет выявить данную патологию у людей после 80 лет. В нее входит:
- Церебральный или мозговой амилоидоз. Диагностируется при болезни Альцгеймера;
- Амилоидоз сердца. Поражает сердечную мышцу. Отложения образуются также в легких, печени и поджелудочной железе.
Опухолевый
В этом случае амилоидоз развивается локально в органах с выраженным злокачественным процессом. Его причиной становится медуллярный рак щитовидной железы или опухоль островков поджелудочной железы.
Гемодиализный
При гемодиализе, назначаемом пациентам с почечной недостаточностью, постепенно повышается содержание в крови B 2 -микроглобулина. Данный белок при взаимодействии с нуклеопротеидами оседает в тканях почек.
Диагностика амилоидоза
Для диагностики амилоидоза пациенту назначается ряд разнообразных исследований. Это общий анализ крови и мочи, биохимия крови, УЗИ внутренних органов, биопсия и генетическое исследование.
Общий анализ крови
Определяет отклонения от нормы, специфические для амилоидоза. На последних стадиях болезни данное исследование помогает выявить поврежденный орган.
Общий анализ мочи
Диагностика амилоидоза почек показывает вероятность развития воспалительных процессов, проходящих в почках.
При патологии почек выявляется:
- Протеинурия – содержание в моче белка свыше 3 г/л;
- Гематурия – обнаружение в моче эритроцитов;
- Лейкоцитурия – присутствие в моче лейкоцитов;
- Цилиндрурия – содержание в моче цилиндров, образующихся при амилоидозе из белков, клеток эпителия почек, лейкоцитов и эритроцитов;
- Снижение плотности мочи.
Биохимический анализ крови
Дает возможность оценить общее состояние организма и установить причину возникновения амилоидоза. При этом анализе определяются:
- Белки общей фазы воспаления, вырабатываемые печенью или определенными лейкоцитами при воспалительном процессе. Особое внимание стоит уделить количеству фибриногена.
- Печеночные пробы указывают на состояние данного органа.
- Увеличение уровня холестерина является признаком нефротического синдрома.
- Снижение уровня белков указывает на нефротический синдром или печеночную недостаточность.
- Повышение количества мочевины и креатинина служит показателем почечной дисфункции при амилоидозе.
Ультразвуковое исследование
Данный метод дает возможность определить структуру и строение тканей внутренних органов, степень и распространение патологических процессов.
Диагностика амилоидоза с помощью УЗИ показывает:
- Уплотнение и изменение размера почек;
- Присутствие кист в почках;
- Уплотнение и увеличение селезенки и печени, сопровождаемое патологией кровотока;
- Гипертрофию сердечной мышцы;
- Наличие отложений амилоида в стенках основных кровеносных сосудов;
- Увеличение объема жидкости в различных полостях организма – асцит, гидроперикард или гидроторакс.
Биопсия
Изъятие небольшой части ткани для исследования с применением специальных методов. Ее использование позволяет диагностировать амилоидоз в 90% случаев заболевания. На исследование берется ткань мышцы, внутренних органов, а также слизистой.
Генетическое исследование
Проводится при вероятности развития наследственного амилоидоза. На исследование берется генетический материал пациента, который проверяется на наличие генетических аномалий в некоторых хромосомах. При обнаружении патологии обследование рекомендуется пройти всем кровным родственникам пациента для выявления у них данного заболевания.
Классификация заболевания почек
Различают первичный и вторичный амилоидоз почек. Первичный амилоидоз характеризуется как заболевание, возникающее в результате неправильного сцепления иммуноглобулинов, которые оседают не только в органах, но и на стенках сосудов, а также поражают состав крови. Более того первичный амилоидоз сложно подается диагностическим процедурам, поскольку имеет большое количество общих симптомов с другими серьезными заболеваниями.
Вторичный амилоидоз развивается в результате хронических инфекций и таких заболеваний как приобретенный остеомиелит, спондилоартрит, колит, заболевания онкологического характера, склероз, ревматоидный артрит и туберкулез. Вторичный амилоидоз может развиться в старческом возрасте, а также как генетически обусловленный фактор.
Всем формам заболевания свойственно:
- возникать и продолжать развиваться преимущественно в период раннего детства;
- протекать с периодическим обострением;
- наличие болевых приступов;
- безрезультативное лечение;
- поражение только почек.
Четыре стадии заболевания:
- Латентная стадия заболевания характеризуется бессимптомным протеканием. Наблюдается лишь отечность и признаки склероза. Стадия может длиться примерно 6 лет. В течение последних 2 лет есть вероятность появления первых признаков основной болезни (туберкулез, артрит, злокачественная опухоль).
- Альбуминурическая стадия заболевания обусловлена появлением амилоид на стенках сосудов. На этой стадии у больного развивается склероз, лимфостаз, а также почки увеличиваются в размерах. Стадия длится примерно 12 лет.
- Отечная стадия. На этой стадии амилоиды полностью поражают нефрон. Основные внутренние органы увеличиваются, отмечается глубокий склероз, выраженная отечность и артериальная гипертензия, наблюдаются признаки очагового поражения кишечного тракта. Стадия длится не более 7 лет.
- Азотемическая стадия – наблюдается сморщенная, деформированная почка, имеющая высокую плотность и рубцы. У больного выражена почечная недостаточность, страдает низким артериальным давлением.
Клиническая картина и прогноз
Амилоидное поражение сердца на ранних стадиях может протекать бессимптомно, проявляясь лишь утолщением стенки ЛЖ при Эхо-КГ [37]. В дальнейшем развиваются клинические проявления СН с застоем крови преимущественно по большому кругу кровообращения [18]. Прогрессирующая одышка, снижение толерантности к физическим нагрузкам отмечаются более чем у половины пациентов и сопровождаются признаками легочной гипертензии [2, 38]. При AL- и старческом ATTR-амилоидозе СН встречается чаще (III–IV функциональные классы по NYHA более чем у половины пациентов), чем при наследственном ATTR-амилоидозе [3, 16]. Системность поражения характерна для AL-амилоидоза. У большинства развиваются поражения почек (нефротический синдром, почечная недостаточность), печени (гигантская гепатомегалия, внутрипеченочный холестаз), селезенки (спленомегалия), мышц (макроглоссия, псевдогипертрофия мышц), кожи (периорбитальная пурпура, амилоидные бляшки), периферической и автономной нервной системы. Изредка развиваются тромбоэмболии коронарных артерий амилоидными массами, приводящие к смерти [39]. Описаны также тромбозы различных сосудов [18].
Диагноз системного амилоидоза устанавливают при гистологическом исследовании ткани, полученной при биопсии. Чаще исследуют стенку прямой кишки, желудка, полости рта, подкожную жировую клетчатку, ткань почки, печени. Определение варианта амилоидоза проводят иммуногистохимическим методом, при наследственном амилоидозе делают анализ ДНК. Если амилоидоз ограничен поражени- ем сердца, как, например, при наследственном ATTR-амилоидозе с мутацией изолейцина в положении 122, то единственным методом диагностики является биопсия миокарда. Однако вбольшинстве других случаев диагноз АС может быть установлен при обнаружении амилоида в биоптате другого органа или ткани и выявлении у больного утолщения стенки ЛЖ более 12 мм по данным Эхо-КГ [40].
Наиболее тяжелый прогноз – при AL-амилоидозе, несколько более благоприятный – при ATTR-амилоидозе. Причем прогноз определяет именно поражение сердца. В исследовании 232 больных AL-амилоидозом с поражением сердца средняя продолжительность их жизни после установления диагноза составила 1 год. Наличие СН снижало выживаемость: 0,75 против 2,34 года у больных без СН.
Традиционно кардиальными предикторами неблагоприятного прогноза помимо тяжести СН и рестриктивных нарушений диастолической функции ЛЖ считают утолщение и увеличение массы ЛЖ со снижением фракции выброса [18, 41]. В то же время C. Rapezzi и соавт. выявили наиболее значительное утолщение стенки миокарда и увеличение массы миокарда у больных старческим ATTR- амилоидозом, при котором отмечается относительно медленное прогрессирование АС [16]. Примечательно, что умеренное утолщение стенок миокарда у больных AL-амилоидозом, участвовавших в этом исследовании, сочеталось со значительным снижением вольтажа желудочковых комплексов ЭКГ. Авторы связали эти изменения на ЭКГ с прямым токсическим эффектом легких цепей иммуноглобулинов на кардиомиоциты. В то же время массивные депозиты амилоида у больных старческим ATTR-амилоидозом, по-видимому, не оказывают столь неблагоприятного действия на клетки миокарда и, следовательно, меньше влияют на выживаемость больных. В другом исследовании 57 больных с AL- и ATTR-амилоидозом низкий вольтаж ЭКГ также сопровождался ухудшением прогноза [42].
В настоящее время важным предиктором неблагоприятного прогноза считают высокие уровни в крови маркеров повреждения кардиомиоцита – тропонинов T и I, N-концевого пробелка мозгового натрийуретического фактора (NT-proBNP) в момент установления диагноза АС. Так, у 261 больного с впервые установленным диагнозом средняя продолжительность жизни при повышении уровня тропонинов I или T составила соответственно 6 и 8 месяцев, а в отсутствие тропонинов I или T в крови – 21 и 22 месяца [43]. В этом исследовании прогностическое значение уровня тропонинов сыворотки было выше, чем значение застойной СН и эхокардиографических параметров. Изучается возможность анализа динамики уровня NT-proBNP после лечения для оценки прогноза АС. При AL-амилоидозе снижение уровня NT-proBNP на 30 % после химиотерапии ассоциировалось с улучшением прогноза [44].
Важным прогностическим фактором являются также наличие и тяжесть внесердечных проявлений системного амилоидоза (ортостатическая гипотония, осложнения нефротического синдрома и моторной диареи, почечная недостаточность и др.).
Таким образом, среди вариантов АС наиболее тяжелым и быстропрогрессирующим течением отличается AL-амилоидоз, при котором развитие СН мало зависит от степени утолщения стенок миокарда и не всегда сочетается с рестриктивными нарушениями диастолической функции. Прогрессирование АС у этих больных в бóльшей степени обусловлено повреждением кардиомиоцита. У больных старческим ATTR-амилоидозом отмечается относительно мягкое течение кардиопатии, несмотря на значительные структурные (выраженную псевдогипертрофию) и функциональные (рестриктивные расстройства гемодинамики) нарушения. Промежуточное положение занимает наследственный ATTR-амилоидоз, при котором симптомы кардиопатии близки к таковым AL-амилоидоза сердца, однако отличаются меньшей злокачественностью.
Нарушения функции сердца
Нарушения внутрисердечной гемодинамики при АС обусловлены главным образом диастолической дисфункцией, снижение сократительной способности миокарда обычно незначительное. В исследовании, включившем 223 больных АС, средняя фракция выброса ЛЖ составила 52,5 ± 13,1 % у больных AL-амилоидозом и 58 ± 13 % у пациентов с наследственным ATTR-амилоидозом. Она была достоверно ниже только среди больных старческим ATTR-амилоидозом 44,2 ± 15,4 % [16]. Снижение фракции выброса менее 40 % было отмечено у 40 % больных старческим ATTR-амилоидозом и только у 22 % пациентов с AL-амилоидозом и 8 % больных наследственным ATTR-амилоидозом. Сходные изменения сократительной функции описывают и другие исследователи [18].
Диастолическая дисфункция разной степени определяется практически для всех пациентов. На ранних этапах развития болезни отложения амилоида нарушают изоволюмическое расслабление миокарда, что приводит к уменьшению скорости раннего диастолического наполнения ЛЖ (E) и увеличению позднего диастолического потока (А). Таким образом, снижение отношения Е/А (1-й тип диастолической дисфункции) является ранним признаком поражения сердца. При прогрессировании АС стенка миокарда становится более жесткой, давление в левом предсердии возрастает, что приводит к увеличению скорости раннего диастолического наполнения и, таким образом, к псевдонормализации отношения Е/А (2-й тип диастолической дисфункции) [2]. При дальнейшем увеличении жесткости сердечной стенки и росте конечного диастолического давления происходит значительное снижение позднего диастолического потока, что позволяет выявить снижение растяжимости миокарда (3-й рестриктивный тип диастолической дисфункции). Рестриктивные изменения часто сопровождаются расширением обоих предсердий [3, 18].
Хотя рестриктивные нарушения диастолической функции считают наиболее типичными для АС, данные исследований показывают возможность развития и других нарушений внутрисердечной гемодинамики [34–36].