Aranesp
Treatment with Aranesp should be carried out by physicians experienced in prescribing for the above indications.
Mode of application
Treatment of symptomatic anemia in combination with chronic renal failure (CRF) in adults and children
Anemia symptoms and consequences may vary depending on patients' age, gender, and severity of the disease; in each case, an analysis of the patient’s individual clinical data by the attending physician is necessary. Aranesp can be used subcutaneously or intravenously to increase hemoglobin levels, but not above 12 g/dL. In patients not on dialysis, the subcutaneous route of administration is preferable, as it avoids puncture of peripheral veins. Patients should be closely monitored to ensure adequate correction of anemia while maintaining hemoglobin concentrations below or at 12 g/dL using the minimum approved doses of Aranesp. The dose of Aranesp should be increased with caution in patients with chronic renal failure. In patients with an insufficient response to Aranesp in terms of hemoglobin levels, alternative explanations for this insufficient response should be considered (see sections "Pharmacodynamics" and "Special Instructions"). Patients' hemoglobin levels are subject to individual fluctuations, including sometimes being higher or lower than desired target values. If the hemoglobin content deviates beyond the target values, the dose is modified, and the target value should be considered to be the range from 10 g/dL to 12 g/dL. Sustained increases in hemoglobin levels above 12 g/dL should be avoided; instructions for dose modification for hemoglobin values above 12 g/dL are provided below. An increase in hemoglobin levels of more than 2 g/dL over a 4-week period should also be avoided. In this case, dose adjustment is also necessary.
Treatment with Aranesp includes two stages - a correction phase and a maintenance phase. Recommendations for use and dosage in adults and children are given separately in the instructions.
Adult patients with chronic renal failure
Correction phase:
The initial dose for subcutaneous or intravenous administration should be 0.45 mcg/kg body weight when administered once weekly. Alternatively, for patients not receiving dialysis, the following initial doses of the drug can also be given subcutaneously: 0.75 mcg/kg body weight every two weeks or 1.5 mcg/kg body weight once a month. If the increase in hemoglobin is not sufficient (less than 1 g/dL for 4 weeks), the dose of the drug must be increased by approximately 25%. Increasing the dose of the drug should not be carried out more often than once every four weeks.
If the increase in hemoglobin exceeds 2 g/dL over 4 weeks, the dose should be reduced by approximately 25%. In cases where the hemoglobin level exceeds 12 g/dL, the possibility of reducing the dose of the drug should be considered. If the hemoglobin level continues to increase, the dose should be reduced by approximately 25%. If, after reducing the dose, hemoglobin continues to increase, it is necessary to temporarily stop using the drug until the hemoglobin level begins to decrease, after which therapy can be resumed, and the dose of the drug should be reduced by approximately 25% of the previous dose.
Hemoglobin levels should be measured weekly or biweekly until they stabilize. Subsequently, the intervals between hemoglobin measurements can be increased.
Maintenance phase:
Patients on dialysis can continue to administer Aranesp once a week or switch to once every two weeks. When switching patients on dialysis from weekly injections of Aranesp to a once-every-two-week regimen, the initial dose should be twice the once-weekly dose.
For patients not receiving dialysis, Aranesp can be continued to be administered once a week or once every two weeks or once a month. For patients receiving Aranesp once every two weeks, once the desired hemoglobin concentration has been achieved, subcutaneous administration of Aranesp may then be given once a month using an initial dose twice the previous biweekly dose.
Dose titration to maintain the required hemoglobin concentration should be done as often as required.
If dose adjustment is necessary to maintain the required hemoglobin concentration, it is recommended to change the dose by approximately 25%.
If the increase in hemoglobin exceeds 2 g/dL over 4 weeks, the dose of the drug should be reduced by approximately 25% depending on the degree of increase in hemoglobin. In cases where the hemoglobin level exceeds 12 g/dL, the possibility of reducing the dose of the drug should be considered. If the hemoglobin level continues to increase, the dose should be reduced by approximately 25%. If, after reducing the dose, hemoglobin continues to increase, it is necessary to temporarily stop using the drug until the hemoglobin level begins to decrease, after which therapy can be resumed, and the dose of the drug should be reduced by approximately 25% of the previous dose.
After any change in dose or administration regimen, hemoglobin levels should be monitored weekly or biweekly. Dose changes during the maintenance phase should be made no more than once every two weeks.
If the route of administration is changed, the same dose of the drug should be used and the hemoglobin level should be monitored once every 1 or 2 weeks to adjust the dose if necessary to maintain the required hemoglobin level.
According to clinical studies, adult patients receiving rhEpo injections once, twice or three times a week can be switched to a once-weekly or biweekly Aranesp regimen. The initial weekly dose of Aranesp (mcg/week) is determined by dividing the total weekly dose of rhEpo (IU/week) by 200. The initial dose of Aranesp * (mcg/two weeks) when administered once every two weeks is determined by dividing the total cumulative dose rhEpo administered over a two-week period by 200. Due to individual differences, selection of the optimal therapeutic dose is required for individual patients. When replacing rhEpo with Aranesp, hemoglobin levels should be measured weekly or every two weeks, and the route of drug administration should remain unchanged.
Children with chronic renal failure
The use of the drug in children under 1 year of age has not been studied in randomized clinical trials (see section “Pharmacodynamics”).
Correction phase:
For children aged 1 year and older, the initial dose for subcutaneous or intravenous administration of the drug is 0.45 mcg/kg body weight as a single injection once a week. In patients not receiving dialysis, an initial dose of 0.75 mcg/kg subcutaneously once every two weeks can be used. If the increase in hemoglobin is not sufficient (less than 1 g/dL for 4 weeks), the dose of the drug must be increased by approximately 25%. Increasing the dose of the drug should not be carried out more often than once every four weeks.
If the increase in hemoglobin exceeds 2 g/dL over 4 weeks, the dose of the drug should be reduced by approximately 25% depending on the degree of increase in hemoglobin. In cases where the hemoglobin level exceeds 12 g/dL, the possibility of reducing the dose of the drug should be considered. If the hemoglobin level continues to increase, the dose should be reduced by approximately 25%. If, after reducing the dose, hemoglobin continues to increase, it is necessary to temporarily stop using the drug until the hemoglobin level begins to decrease, after which therapy can be resumed, and the dose of the drug should be reduced by approximately 25% of the previous dose.
Hemoglobin levels should be measured weekly or biweekly until they stabilize. Subsequently, the intervals between hemoglobin measurements can be increased.
Treatment of anemia with Aranesp in the correction phase in children in a once-a-month dosing regimen has not been studied.
Maintenance phase:
In children aged 1 year and older, during the maintenance phase of therapy, the administration of Aranesp can be continued once a week or once every two weeks. Patients under 6 years of age may require a higher dose to maintain hemoglobin levels than older patients. When switching patients on dialysis from weekly injections of Aranesp to a once-every-two-week regimen, the initial dose should be twice the once-weekly dose.
For patients aged 11 years and older who are not on dialysis, after the target hemoglobin level has been achieved with a biweekly dosing regimen, Aranesp can be administered subcutaneously once a month, with the initial dosage being twice that dose. , which was used in the once every two weeks mode. Clinical data have shown that pediatric patients receiving rhEPO two or three times a week can be switched to Aranesp once weekly, and patients receiving rhEPO once a week can be switched to Aranesp once every two weeks. . The starting dosage of Aranesp for children (mcg/week) administered weekly can be determined by dividing the total weekly dose of rhEpo (IU/week) by 240. The starting dosage of Aranesp when administered every 2 weeks (mcg/every 2 weeks) can be determined by dividing the total dose of rhEpo over a two-week period by 240. Due to individual differences, selection of the optimal therapeutic dose is required for individual patients. When replacing rhEpo with Aranesp, hemoglobin levels should be measured weekly or every two weeks, and the route of drug administration should remain unchanged.
Dose titration to maintain the required hemoglobin concentration should be done as often as required.
If dose adjustment is necessary to maintain the required hemoglobin concentration, it is recommended to change the dose by approximately 25%.
If the increase in hemoglobin exceeds 2 g/dL over 4 weeks, the dose of the drug should be reduced by approximately 25% depending on the degree of increase in hemoglobin. In cases where the hemoglobin level exceeds 12 g/dL, the possibility of reducing the dose of the drug should be considered. If the hemoglobin level continues to increase, the dose should be reduced by approximately 25%. If, after reducing the dose, hemoglobin continues to increase, it is necessary to temporarily stop using the drug until the hemoglobin level begins to decrease, after which therapy can be resumed, and the dose of the drug should be reduced by approximately 25% of the previous dose.
In patients starting dialysis while on Aranesp therapy, hemoglobin levels should be carefully monitored.
After any change in dose or administration regimen, hemoglobin levels should be monitored weekly or every two weeks. Dose changes during the maintenance phase should be made no more than once every two weeks.
If the route of administration of the drug is changed, the same dose of the drug should be used and the hemoglobin level should be monitored once every 1 or 2 weeks to adjust the dose if necessary in order to maintain the required hemoglobin level.
Treatment of symptomatic chemotherapy-induced anemia in patients with cancer
In patients with anemia (eg, with a hemoglobin concentration equal to or less than 10 g/dL), Aranesp can be used subcutaneously to increase the hemoglobin concentration to a level not exceeding 12 g/dL. Anemia symptoms and consequences may vary depending on patients' age, gender, and severity of the disease; in each case, an analysis of the patient’s individual clinical data by the attending physician is necessary. Patients' hemoglobin levels are subject to individual fluctuations, including sometimes being higher or lower than desired target values. If the hemoglobin content deviates beyond the target values, the dose is modified, and the target value should be considered to be the range from 10 g/dL to 12 g/dL. Increases in hemoglobin levels above 12 g/dL should be avoided; Below is a guideline for dosage adjustments if hemoglobin levels exceed 12 g/dL.
The recommended initial dose of the drug is 500 mcg (6.75 mcg/kg) once every 3 weeks or 2.25 mcg/kg once a week. If the clinical response (fatigue, hemoglobin level) is inadequate after nine weeks, further therapy may not be effective.
Aranesp is discontinued approximately four weeks after completion of chemotherapy.
After achieving the target hemoglobin level, the dosage of the drug should be reduced by 25-50% to adequately control the symptoms of anemia using the minimum approved doses of Aranesp. It is possible to titrate the dose between 500 mcg, 300 mcg and 150 mcg.
Patients should be closely monitored. If the patient's hemoglobin level exceeds 12 g/dL, the dose of the drug should be reduced by 25-50%. If the hemoglobin level exceeds 13 g/dL, use of Aranesp should be temporarily discontinued. After the hemoglobin level has decreased to 12 g/dL or lower, therapy can be resumed, the dosage of the drug should be approximately 25% less than the previous one.
If the increase in hemoglobin exceeds 2 g/dL over 4 weeks, the dose of the drug should be reduced by approximately 25-50%.
Method of administration
Aranesp can be administered subcutaneously by the patient or caregiver after training by the attending physician, nurse, or pharmacist.
The drug Aranesp at a dosage of 20 mcg, 30 mcg, 500 mcg in the dosage form, solution for injection in a pre-filled syringe (PZS)
Aranesp can be administered subcutaneously or intravenously as described in the dosage regimen.
It is necessary to change injection sites and administer the drug slowly to avoid discomfort at the injection site.
Aranesp is supplied ready for use in pre-filled syringes. Instructions for using the drug, handling it and the procedure for its destruction are given in the “Special Instructions” section.
Pharmaceutical incompatibility
Since compatibility studies have not yet been conducted, this medicinal product should not be mixed or infused with other medicinal products.
TECHNIQUE FOR INJECTION OF ARANESP IN PRE-FILLED SYRINGES
This section describes the procedure for injecting Aranesp, which you can do yourself. Before using the drug in pre-filled syringes, please first read the “General Recommendations” below (Section 1) and then the instructions (Section 2).
Section 1. General recommendations
It is very important that you do not give the injection yourself until your doctor, nurse or pharmacist teaches you to do so. If you have any questions, consult your doctor, nurse or pharmacist.
Before starting the injection
Read all recommendations carefully before administering the drug.
How should you, or the person giving you this injection, use Aranesp PZSh?
Your doctor has prescribed Aranesp PZSh for subcutaneous injection. Your doctor, nurse or pharmacist will tell you how much Aranesp should be administered and how often.
Equipment:
For self-injections you will need:
— New PZSh with the drug Aranesp and
— Alcohol-soaked swabs or similar materials.
Before the injection
1. Remove the prefilled syringe from the refrigerator. Leave the PZSh at room temperature for about 30 minutes. This will make the injection more comfortable. Do not heat the prefilled syringe in any other way (for example, in hot water or in a microwave oven). Do not leave the pre-filled syringe in direct sunlight.
2. Do not shake the pre-filled syringe.
3. Do not remove the gray cap of the prefilled syringe until you are ready to inject.
4. You use the drug and dose prescribed by your doctor.
5. The expiration date on the label of the pre-filled syringe (BEAR BEFORE). Do not use PZSh if the last day of the specified month has expired.
6. Description of the drug Aranesp. The solution should be clear, colorless or light yellow. If the solution is cloudy or contains particles, the drug should not be used.
7. Wash your hands thoroughly.
8. Choose a comfortable, well-lit place and a clean surface, conveniently arrange all the necessary materials.
How to choose an injection site?
It is best to inject into the upper thigh and abdomen. If someone else is giving you the injections, you can use the outside of your upper arm.
If the area you are about to inject is red or swollen, you can choose a different injection site.
Section 2. Recommendations for administering Aranesp in a pre-filled syringe
How to prepare for an injection with Aranesp?
Before injection with Aranesp, you must do the following:
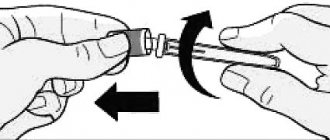
1. To avoid bending the needle, carefully pull the cap off the needle immediately without twisting, as shown in Figures 1 and 2.
2. Do not touch the needle or press the plunger.
3. You may notice small air bubbles in the prefilled syringe. You do not need to remove air bubbles before injection. Injecting the solution with air bubbles is safe.
4. You can now use the pre-filled syringe.
How to administer the drug?
1. Disinfect the injection site using a swab soaked in alcohol, and pinch the skin (without squeezing) with your thumb and forefinger.
2. Insert the needle completely into the skin as indicated by the doctor, nurse or pharmacist.
3. Administer the prescribed dose subcutaneously, as directed by your doctor, nurse or pharmacist.
4. Slowly and continuously press the plunger, while squeezing the skin fold and do not release it until the syringe is empty.
5. Remove the needle and release the skin fold.
6. If blood comes out, gently wipe it off with a cotton ball or cloth. Do not rub the injection site. If necessary, you can seal it with adhesive tape.
7. One pre-filled syringe is for single use. Do not use Aranesp remaining in the syringe.
Remember
: If difficulties arise, seek help or advice from your doctor or nurse.
Aranesp
Aranesp
RLS > Medicines and substances > Aranesp Aranesp (Aranesp) Latest update of the description by the manufacturer 10/12/2012 Aranesp 10/12/2012 Active ingredient: Darbepoetin alfa* (Darbepoetin alfa*) Contents ATC B03XA02 Darbepoetin alfa Pharmacological group Nosological classification (ICD-10) Composition Solution for injection 1 pre-filled syringe active substance: darbepoetin alfa (recombinant) - 10 mcg (25 mcg/ml); 15 mcg (40 mcg/ml); 20 mcg (40 mcg/ml); 30 mcg (100 mcg/ml); 40 mcg (100 mcg/ml); 50 mcg (100 mcg/ml); 60 mcg (200 mcg/ml); 80 mcg (200 mcg/ml); 100 mcg (200 mcg/ml); 150 mcg (500 mcg/ml); 300 mcg (500 mcg/ml); 500 mcg (500 mcg/ml) excipients in 1 ml of solution: sodium dihydrogen phosphate monohydrate - 2.118 mg; sodium hydrogen phosphate - 0.661 mg; sodium chloride - 8.182 mg; polysorbate 80 - 0.05 mg; water for injection - up to 1 ml Description of the dosage form Transparent, colorless liquid. Characteristics Darbepoetin alfa is produced using gene technology in Chinese hamster ovary cells (CHO-K1). Pharmacological action Pharmacological action - hematopoietic. Pharmacodynamics Darbepoetin alfa stimulates erythropoiesis by the same mechanism as endogenous erythropoietin. Darbepoetin alfa contains 5 N-linked carbohydrate chains, while the endogenous hormone and recombinant human erythropoietin (rhEPO) have only three chains. Additional sugar residues, from a molecular point of view, do not differ from those present in the endogenous hormone. Due to its increased carbohydrate content, darbepoetin alfa has a longer half-life compared to rhEPO and, therefore, greater activity in vivo. Despite these changes in molecular structure, darbepoetin alfa retains a very narrow specificity for the erythropoietin receptor. Erythropoietin is a growth factor that primarily stimulates the formation of red blood cells. Erythropoietin receptors can be expressed on the surface of various tumor cells. Patients with chronic renal failure In patients with chronic renal failure, 2 clinical studies found that the risk of death and serious cardiovascular adverse events was higher when erythropoiesis-stimulating agents were used to higher target hemoglobin levels compared with lower target levels of 135 g/L (8 .4 mmol/l) versus 113 g/l (7.1 mmol/l); 140 g/l (8.7 mmol/l) versus 100 g/l (6.2 mmol/l). In the randomized, double-blind, placebo-controlled trial (TREAT), 4038 patients with chronic renal failure, type 2 diabetes and hemoglobin levels ≤110 g/L not on dialysis received darbepoetin alfa to achieve a hemoglobin level of 130 g/L or placebo ( with the appointment of darbepoetin alfa when the hemoglobin level decreases below 90 g/l). The study did not meet the primary goal of reducing the risk of all-cause mortality or cardiovascular morbidity (darbepoetin alfa vs placebo; hazard ratio 1.05; 95% CI (0.94, 1.17), nor did it meet the goal of in reducing all-cause mortality and progression to end-stage renal disease (ESRD) (darbepoetin alfa vs placebo; hazard ratio 1.06; 95% CI (0.95, 1.19). Analysis of individual components of composite endpoints showed the following ratio risks (95% CI): death 1.05 (0.92; 1.21), chronic heart failure (CHF) 0.89 (0.74; 1.08), myocardial infarction (MI) 0.96 ( 0.75; 1.23), stroke 1.92 (1.38; 2.68), hospitalization due to myocardial ischemia 0.84 (0.55; 1.27), ESRD 1.02 (0.87 1,18 Cancer patients receiving chemotherapy Survival and tumor progression were studied in a total of 2833 patients in five large controlled studies, four of which were double-blind and placebo-controlled, and one was open-label. Two studies included patients who had already received chemotherapy treatment. Two studies set the target hemoglobin level at or above 130 g/L, and three others set the target hemoglobin level between 120 and 140 g/L. In an open-label study, there was no difference in overall survival between the rhEPO-treated group and the control group. In four placebo-controlled studies, risk ratios favored the control group and ranged from 1.25 to 2.47. These four studies found an unexplained, statistically significant increase in mortality compared with controls in patients with typical cancers and anemia treated with rhEPO. Comparison of the incidence of thrombosis and other complications in the groups treated with rhEPO and the control group does not provide a satisfactory explanation of the reasons for this increase. A systematic analysis of 57 studies was also conducted, including a total of more than 9,000 cancer patients. In a meta-analysis of overall survival, the hazard ratio was 1.08 in favor of controls (95% CI: 0.99–1.18; 8167 patients in 42 studies). Patients treated with rhEPO had an increased relative risk of thromboembolic events (RR=1.67; 95% CI: 1.35–2.06; 6769 patients in 35 studies). In summary, there is sufficient evidence to suggest that significant harm may occur when treating cancer patients with rHuEPO. It is unclear to what extent this applies to the use of recombinant human erythropoietins to achieve a hemoglobin target of less than 130 g/L in cancer patients receiving chemotherapy, as the data analyzed included a small number of patients with these characteristics. We also analyzed data from more than 13,900 patients with malignant disease (chemotherapy, radiotherapy, chemotherapy and radiation therapy or no therapy) included in 53 controlled clinical trials of several epoetins. A meta-analysis of overall survival data found a hazard ratio of 1.06 in favor of the control group (95% CI: 1, 1.12; 53 studies and 13,933 patients), and for patients with cancer receiving chemotherapy, the overall survival hazard ratio was 1.04 (95% CI: 0.97, 1.11; 38 studies and 10,441 patients). The meta-analysis also indicates a significant increase in the relative risk of thromboembolic events in patients with malignancies receiving recombinant human erythropoietin (see section "Special Instructions"). Preclinical safety data In all studies in rats and dogs, darbepoetin alfa significantly increased the concentration of hemoglobin, hematocrit, red blood cells and reticulocytes, which corresponds to the expected pharmacological effect. Adverse events when administering very high doses of the drug were considered as a consequence of enhanced pharmacological action (decreased tissue blood flow due to increased blood viscosity). This also included myelofibrosis and splenic hypertrophy, as well as widening of the QRS complex on the ECG in dogs, without disturbing the heart rhythm and affecting the QT interval. Darbepoetin alfa did not have any genotoxic potential and did not affect the proliferation of non-hematological cells either in vitro or in vivo. In chronic toxicity studies, no tumorigenic or unexpected mitogenic response was observed in any tissue type studied. The carcinogenic potential of darbepoetin alfa has not been assessed in long-term animal studies. In trials conducted in rats and rabbits, no clinically significant effects on pregnancy, embryonic/fetal development, parturition or postnatal development were observed. The level of penetration of the drug through the placenta was minimal. No changes in fertility were noted. Pharmacokinetics Due to the increased carbohydrate content, the concentration of darbepoetin alfa circulating in the blood exceeds the minimum concentration required to stimulate erythropoiesis for a longer time, compared with equivalent doses of rhEPO, which allows reducing the frequency of administration of darbepoetin alfa while maintaining an equivalent level of biological response. Patients with chronic renal failure The pharmacokinetics of darbepoetin alfa have been studied in patients with chronic renal failure with intravenous and subcutaneous administration of the drug. Its elimination half-life was 21 hours (standard deviation (SD 7.5)) when administered intravenously. The clearance of darbepoetin alfa was 1.9 ml/h/kg (SD 0.56), and the volume of distribution (Vd) was approximately equivalent to the volume of plasma (50 ml/kg). With subcutaneous administration of the drug, its bioavailability corresponded to 37%. With monthly subcutaneous administration of darbepoetin alfa at a dose of 0.6 to 2.1 mcg/kg, its half-life was 73 hours (SD 24 The longer half-life of darbepoetin alfa when administered subcutaneously compared with intravenous administration is due to absorption kinetics. In clinical studies, minimal drug accumulation was observed with any route of administration. In preclinical studies, renal clearance of darbepoetin was demonstrated to be minimal ( up to 2% of total clearance) and does not affect the serum half-life of the drug.The pharmacokinetics of darbepoetin alfa was studied in children (3-16 years) with chronic renal failure, on dialysis or not, with samples taken from the moment of a single s.c. or intravenous administration of the drug up to one week (168 hours) after administration. Sampling periods were of similar duration as in adults with chronic renal failure, and the comparison showed that the pharmacokinetics of darbepoetin alfa were similar in adults and children with chronic renal failure. After intravenous administration of the drug, there was an approximately 25% difference between adults and children in terms of the area under the concentration-time pharmacokinetic curve from time zero to infinity (AUC0-∞); however, the reported difference for children was less than 2-fold of the AUC0-∞ range. After subcutaneous administration of the drug, the AUC0-∞ value in adults and children was similar. Both after intravenous and subcutaneous administration of the drug, the half-life of the drug in children and adults with chronic renal failure was similar. Cancer patients receiving chemotherapy After subcutaneous administration of the drug at a dose of 2.25 mcg/kg to adult cancer patients, the mean maximum concentrations (Cmax) of darbepoetin alfa, amounting to 10.6 ng/ml (SD 5.9), were established on average in for 91 hours (SD 19.7). These parameters corresponded to linear pharmacokinetics over a wide range of values (from 0.5 to 8 mcg/kg when administered weekly and from 3 to 9 mcg/kg when administered once every 2 weeks). Pharmacokinetic parameters were not affected by repeated dosing over 12 weeks (weekly or biweekly dosing). There was an expected moderate increase (<2-fold) in serum concentrations of the drug upon reaching steady state, but there was no evidence of accumulation upon repeated administration. Pharmacokinetic studies were performed in patients with chemotherapy-induced anemia who received darbepoetin alfa injections at a dose of 6.75 mcg/kg every three weeks in combination with SC chemotherapy. In this study, the mean (SD) half-life was 74 (SD 27) hours. This video introduces Aranesp. Indications for the drug Aranesp are the treatment of symptomatic anemia in adults and children suffering from chronic renal failure (CRF); therapy of symptomatic anemia in adult cancer patients with non-myeloid malignancies receiving chemotherapy.