Throughout life, a person repeatedly experiences abdominal pain. Under the age of 50, most often the cause of pain is not organic, but functional in nature, when drug therapy is most likely to be effective. The cause may be intestinal colic (in children in the first months of life), irritable bowel syndrome, biliary dyskinesia, dysfunction of the sphincter of Oddi, etc. About a third of cases of functional pain are accompanied by spasm of the smooth muscles of the gastrointestinal tract. This is why antispasmodics are so widely used without a doctor’s prescription. Providing a pronounced analgesic effect and normalizing the functioning of the organ, they, unlike non-narcotic and opioid analgesics, do not interfere with the mechanisms of pain, that is, they do not “erase” the picture of the disease, which is critically important in diagnosis.
Nota bene!
When a client asks for a remedy for abdominal pain, it is necessary to exclude urgent situations in order to send the sufferer to a doctor in time. These include:
- Severe pain that prevents you from sleeping or doing anything, lasts longer than 1-2 hours.
- Severe abdominal pain is accompanied by vomiting.
- Severe pain is accompanied by elevated body temperature - 38.5°C and above.
- Severe pain is accompanied by loss of consciousness.
- Severe abdominal pain in a pregnant woman.
- The abdominal muscles are tense and the stomach is as hard as a board.
- Diarrhea mixed with bright red blood.
- The stool is dark and tarry.
- Vomiting blood.
- Abdominal pain is accompanied by vomiting, diarrhea and severe dehydration.
Cellular mechanism of pain
Smooth muscle cells are mainly found in the large intestine. There are significantly fewer of them in the small intestine. The mechanism of cell contraction depends on the concentration of calcium ions in the cytoplasm. The sources of calcium are the extracellular space, connected to the cytoplasm through calcium (slow) membrane channels, as well as the intracellular depot. The release of calcium causes the muscle cell to contract. The contraction of muscle cells forms a spasm and the person feels pain.
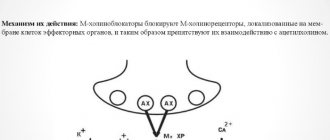
The process of muscle contraction is most often triggered by the mediator acetylcholine, binding to muscarinic cholinergic receptors (M-cholinergic receptors). Thus, blockade of M-cholinergic receptors leads to a decrease in tone and relaxation of smooth muscles (and therefore helps relieve abdominal pain). This, in particular, is the basis for the mechanism of action of a group of drugs called M-anticholinergics.
The interaction of calcium with calmodulin leads to the interaction of actin with myosin and cell contraction. CAMP (cyclic adenosine monophosphate) and cGMP (cyclic guanosine monophosphate) weaken the interaction of calcium with calmodulin and also impede the entry of calcium ions into the cell. The level of cAMP and cGMP is controlled by phosphodiesterase (PDE), which destroys excess of these nucleotides. Thus, if you reduce the activity of PDE, the concentration of calcium ions in the cytoplasm will decrease and the tone of the muscle cell will decrease.
Based on the described mechanisms for regulating the calcium concentration inside the muscle cell, which are disrupted under pathological conditions and lead to spasm, the main groups of antispasmodics have been created:
- M-anticholinergics (atropine, hyoscine butyl bromide).
- PDE IV inhibitors (drotaverine).
- Blockers of calcium release from intracellular stores or sodium channel blockers (mebeverine).
- Calcium channel blockers.
General information
Anticholinergics refer to medications that block and weaken the action of the neurotransmitter responsible for the occurrence of excitation of the nervous system. Pharmacological agents from this group can affect different structures of the body.
Alkaloids are the most common anticholinergics:
- Scopolamine
- Platyfillin
- Atropine
- Plant based medicines
Today, synthetic substances are often used. Among their components there are elements that well enhance the effect of anticholinergics. They are very convenient to use, negative reactions of the body are reduced.
Many anticholinergics are excellent as analgesic and antispasmodic agents.
This group of drugs has properties inherent in antihistamine and local anesthetic drugs. Diprazine and Diphenhydramine are among the most popular medications of this subtype. It is worth considering that the structure of pharmaceutical anticholinergic drugs is different from each other.
Anticholinergic drugs are divided into:
- N-anticholinergics
- M-anticholinergics
M-anticholinergics
M-anticholinergics are one of the oldest groups of drugs.
Atropine has been used in medicinal herbs since the 4th century BC. e. Thus, the ancient Greek naturalist Theophrastus described mandrake as a remedy for treating wounds, gout and insomnia, and also as a “love potion.” The ancient Greek military physician Dioscorides (1st century AD) described mandrake wine as an anesthetic to treat pain or insomnia, to be given before surgery or cauterization. M-anticholinergics most effectively affect the upper gastrointestinal tract. The use of non-selective M-anticholinergics (atropine, platyphylline, metacin) is limited by their prescription, as well as by the fact that they have a systemic effect and have a number of side effects, such as intestinal atony and obstruction.
One of the relatively oldest selective M-anticholinergics is hyoscine butyl bromide (first registered in Germany in 1951). It acts on the M1 and M3 subtypes of receptors, localized mainly in the walls of the upper gastrointestinal tract, gallbladder and biliary ducts. Hyoscine butyl bromide does not penetrate the blood-brain barrier and has a weak systemic effect, unlike atropine. However, it is contraindicated in people with glaucoma, benign prostatic hyperplasia, as well as organic stenosis of the gastrointestinal tract and tachyarrhythmias. Hyoscine butyl bromide accumulates in the smooth muscles of the gastrointestinal tract and is excreted unchanged by the kidneys. The drug is successfully used for sphincter of Oddi dysfunction, biliary dyskinesia and pyloric spasm.
The severity of the effect of hyoscine butylbromide also depends on the patient’s individual sensitivity to this drug. Unfortunately, the antispasmodic effect of hyoscine butyl bromide on the small intestine (except the duodenum) and large intestine can be realized when taking a dose 2–10 times higher than the therapeutic dose, so its use is useless for spasms in the lower abdomen. A large number of restrictions and risks when taking this drug require medical supervision, which is why the M-anticholinergics known to us are currently used less and less abroad. However, hyoscine butyl bromide is still available without a prescription.
Antispasmodics: from clinical pharmacology to pharmacotherapy
Drugs belonging to the group of antispasmodics are among the most popular and prescribed by doctors of various specialties. Dysfunction of smooth muscle cells of internal organs underlies both chronic and acute pain syndromes in various diseases of the digestive system and urinary system of the pelvic organs [1, 2]. The most common cause of pain in pathologies of internal organs that have a layer of smooth muscle cells is their excessive contraction. Symptoms of spastic dysfunction of the digestive organs occur in 30% of cases in the absence of organic damage to the stomach or intestines, which leads to the frequent independent (without medical prescription) use of antispasmodic drugs. The widespread use of drugs in this group dictates the need to constantly remind about their clinical pharmacology, especially properties and tolerability, in order to avoid both underestimation and overestimation of therapeutic capabilities.
It is important that, by eliminating or preventing spasms of smooth muscles, antispasmodics, although they have a pronounced analgesic effect, normalizing the functioning of the organ, do not interfere with the mechanisms of pain. Unlike non-narcotic and opioid analgesics, they do not pose a significant threat of “erasing” symptoms in severe organic damage (masking the picture and making it difficult to verify the diagnosis).
Antispasmodics are most often used for:
- symptomatic treatment if spasm is a characteristic accompanying symptom of the disease, but does not play a role in pathogenesis;
- etiotropic therapy if spasm underlies the pathological condition;
- premedication when preparing patients for various procedures.
The process of contraction of a muscle cell is determined by the concentration of calcium ions in its cytoplasm, entering from the extracellular space through calcium (slow) channels of cell membranes. Another source of calcium is intracellular depots, which are more present in the smooth muscles of the colon and much less in the small intestine, including the duodenum. The release of this fraction of calcium ions leads to a phase contraction of the muscle cell [3]. One of the most common mediators influencing the process of muscle contraction is acetylcholine. When it interacts with muscarinic cholinergic receptors (M-cholinergic receptors), sodium channels open, sodium ions enter the cell and, as a result, membrane depolarization occurs. Depolarization, in turn, leads to the opening of calcium channels and the entry of calcium into the cell, increasing its intracellular concentration and muscle contraction. Activation of M-cholinergic receptors by acetylcholine leads to an increase in tone and contraction of smooth muscle cells, and blockade leads to a decrease in tone and relaxation of smooth muscle cells, which underlies the prescription of M-cholinergic blockers as antispasmodics [4, 5].
Calcium interacts with calmodulin, activates myosin light chain kinase, which cleaves the phosphorus residue from the ATP molecule bound to myosin, which leads to the interaction of actin and myosin and cell contraction. The most important role in muscle functioning is known to be played by the system of cyclic nucleotides [6]. Cyclic adenosine monophosphate (cAMP) and cyclic guanosine monophosphate (cGMP) inhibit the release of intracellular calcium and the entry of calcium ions into the cell (cAMP), weaken the interaction of calcium ions with calmodulin. The levels of cAMP and cGMP are controlled by phosphodiesterase (PDE), which breaks down these nucleotides. Thus, the higher the PDE activity, the lower the concentration of cAMP and cGMP, the higher the concentration of calcium ions in the cytoplasm and the tone of the muscle cell and, conversely, the lower the PDE activity, the higher the concentration of cAMP and cGMP, the lower the concentration of calcium ions in the cytoplasm and muscle cell tone. The physiological role of PDE is diverse. 11 isoforms of this enzyme have been identified [4]. The main types of PDEs that affect the tone of smooth muscle cells are presented in
.
The mechanisms described above for regulating the calcium concentration inside the muscle cell, which are disrupted under pathological conditions and lead to spasm of the smooth muscle cells of the internal organs, imply the presence of several targets for pharmacological action [7]:
- blockade of neurotropic effects, that is, blocking the effect of acetylcholine on the M-cholinergic receptor (atropine, hyoscine butyl bromide);
- inhibition of PDE IV activity (drotaverine (No-spa));
- blockade of calcium ion entry (calcium channel blockers);
- blockade of calcium release from intracellular stores (mebeverine).
M-anticholinergics
This is one of the oldest groups of drugs used in medicine to affect the upper gastrointestinal tract. They are 5–10 times more effective in affecting the motility of the stomach than the large intestine, which is associated with the unequal density of M-cholinergic receptors in different parts of the gastrointestinal tract [8]. The largest number of receptors is in the stomach, much less in the intestines. The classic anticholinergic atropine has lost its value due to a large number of side effects. An attempt to increase the safety of therapy with M-anticholinergics was the creation of hyoscine butyl bromide, which is a relatively selective M-anticholinergic blocker acting on the M1 and M3 receptor subtypes, localized primarily in the walls of the upper gastrointestinal tract, gall bladder and biliary ducts. Unlike atropine, the drug does not penetrate the blood-brain barrier and has low (8–10%) systemic bioavailability, which leaves its mark on the variability in the manifestations of the effect.
Hyoscine butyl bromide accumulates in the smooth muscles of the gastrointestinal tract and is excreted unchanged from the body by the kidneys. It acts mainly on the stomach, duodenum, and gall bladder. The antispasmodic effect on the small and large intestines is realized in doses 2–10 times higher than therapeutic ones, therefore, for spasms of these parts of the intestine, use is not advisable (it is better to use myotropic antispasmodics, for example, drotaverine (No-shpa)) [9].
Anticholinergics are successfully used for abdominal pain caused by spasm in the upper gastrointestinal tract: sphincter of Oddi dysfunction, biliary dyskinesia, pylorospasm. Despite the fact that hyoscine butylbromide does not penetrate the central nervous system, it can cause side effects typical of M-anticholinergics (4% more often than placebo). This makes it, like other drugs in the group, contraindicated for glaucoma, benign prostatic hyperplasia, organic stenosis of the gastrointestinal tract, and tachyarrhythmias [10]. Since the drug is sold without a prescription and may cause unwanted effects in patients suffering from these diseases, it is important to inform them in advance. Unfortunately, the importance of selectivity of drug action is often unreasonably exaggerated. Thus, the statement that hyoscine butylbromide acts only at the site of spasm seems controversial, since M-cholinergic receptors are not located locally.
From the point of view of the breadth of effects on organs and systems of hyoscine, butyl bromide is no less effective than myotropic drugs (drotaverine (No-shpa)), but its action, unlike myotropic drugs, can increase the risks of its use: it should not be taken by children under 6 -years of age, simultaneously with antidepressants (especially tricyclics), antihistamines and antiarrhythmic drugs, as well as beta-agonists (high risk of negative interaction between these drugs). Hyoscine butylbromide, like other M-anticholinergics, worsens the condition of patients with reflux esophagitis, hiatal hernia due to relaxation of the lower esophageal sphincter and increased reflux of acidic contents into the esophagus. In elderly patients with chronic atonic constipation, in weakened patients, there is a risk of developing intestinal obstruction. In case of chronic lung diseases, when using M-anticholinergics, thickening of secretions may occur and the degree of bronchial obstruction may increase. In men, a decrease in potency is possible.
With prolonged use, the drug should be discontinued gradually; during treatment, it is strictly forbidden to drink alcoholic beverages, be in the hot sun, perform intense physical work or play sports (impairment of the functions of the sweat glands by the drug with simultaneous insolation can lead to heat stroke).
Thus, the status of hyoscine butyl bromide as an over-the-counter drug does not relieve the patient from the need for periodic medical monitoring. It is precisely these problems that are responsible for the fact that currently the M-anticholinergics known to us abroad are used less and less often. In addition, the severity of their effect depends on the initial tone of the parasympathetic nervous system, which determines significant individual differences in the effectiveness of drugs in patients with varying severity of vagotonia.
Phosphodiesterase inhibitors (PDEs)
The effect of traditional widely used antispasmodics, such as drotaverine (No-shpa), is based on the suppression of PDE IV activity. Since PDE IV is present in smooth muscle cells along the entire length of the intestine, biliary and urinary tracts, its blockade with drotaverine (No-shpa) has a universal antispasmodic effect, regardless of the degree of contraction or the cause that caused it. Drotaverine, unlike hyoscine butylbromide and other myotropic antispasmodics, has anti-edematous and anti-inflammatory effects (type IV PDE is actively involved in the development of inflammation).
Pharmacodynamic properties underlie the pathogenetic therapeutic effect of drotaverine and are widely used not only to relieve acute spastic syndrome, but also to provide long-term pharmacotherapy, for example, with biliary dyskinesias, cholelithiasis, chronic intestinal diseases with spastic syndrome accompanied by colicky or bursting pain. The absence of anticholinergic activity has a positive effect on the safety of drotaverine, expanding the range of people to whom it can be prescribed, in particular, restrictions on use in children are lifted (possible from the age of one year), in elderly men with prostate pathology, as well as with concomitant pathology and when taken together with other drugs.
The therapeutic concentration of drotaverine in plasma when taken orally is observed within 45 minutes. After a single oral dose of 80 mg, maximum plasma concentration is achieved after 2 hours, and oral bioavailability is 60%. The drug penetrates well into various tissues, is metabolized (oxidized) almost completely to monophenolic compounds, the metabolites are quickly conjugated with glucuronic acid. The half-life is 16 hours. About 60% of drotaverine when taken orally is excreted through the gastrointestinal tract and up to 25% in the urine. The presence of a dosage form for both enteral and parenteral administration makes it possible to widely use the drug in an emergency situation, followed by a transition to oral administration for long-term use. Thus, parenteral administration of drotaverine (No-shpa) provides a quick and strong antispasmodic effect, which is especially important in the development of acute, intense colicky spastic pain.
Drotaverine (No-shpa, No-shpa forte) is effective in the treatment of various gastrointestinal diseases characterized by spasm of smooth muscles (biliary dyskinesia, spasm of the sphincter of Oddi, pylorospasm, irritable bowel syndrome), is actively used for urolithiasis, increased blood pressure, disorders of regional blood flow, including for self-medication.
In a double-blind, placebo-controlled clinical trial, the use of No-shpa significantly reduced pain in sphincter of Oddi dyskinesia in 60% of patients, while placebo did not affect the intensity of pain in 55% of cases. In this study, No-shpa turned out to be 2 times more effective than analgesics. The research results showed that No-Spa is the drug of choice for spasms of the smooth muscles of the biliary system, both for monotherapy and in combination with other drugs and surgical methods of treatment. In a double-blind, randomized, placebo-controlled study in 62 patients with irritable bowel syndrome and constipation, abdominal pain (by 47%) and flatulence significantly decreased during 8 weeks of treatment with No-shpa. In another randomized, double-blind study, the effectiveness of No-shpa was studied in 70 aged patients with irritable bowel syndrome for 4 weeks. The results showed that No-shpa significantly reduces the frequency of pain compared to placebo - by 47% (p < 0.001), flatulence by 21% and dyspepsia - by 20% (p < 0.001), improves passage time in the colon [11] .
Long-term use of the drug is safe, which is associated with the absence of an effect on cholinergic structures and significantly improves its tolerability. No-spa is included in the standards for the diagnosis and treatment of diseases of the digestive system approved in the Russian Federation.
Sodium channel blockers
The action is based on disrupting the depolarization of the membrane of smooth muscle cells of the gastrointestinal tract, reducing the permeability of the membrane to extracellular sodium and, in addition, indirectly suppressing the release of potassium from the cell. The result of this is a disruption of the depolarization process and blocking of intracellular calcium stores. The most well-known drug is the veratric acid derivative mebeverine. It binds to the molecular structures of sodium channels without affecting the cholinergic receptor, reduces the outflow of ionized potassium from the cell, and therefore eliminates spasm without causing hypotension of the colon [12]. Metabolized when passing through the intestinal wall and liver, drug metabolites are excreted in the urine.
Mebeverine is prescribed mainly for functional diseases of the gastrointestinal tract (non-ulcer dyspepsia, irritable bowel syndrome), as well as for secondary spasms caused by organic diseases of the intestines and biliary tract. It is used exclusively as planned, i.e., only a course of treatment is provided, unlike drotaverine, which can be used both for the relief and prevention of spastic pain.
The effectiveness of mebeverine is not always observed with monotherapy. Thus, in an extensive pharmacoepidemiological study [13], it was shown that in irritable bowel syndrome in comparison with control (standard therapy), it requires more joint drug support than in control, which leads to increased costs (Table 2).
From the table 2 it is clear that the odds ratio is always higher in the mebeverine group, which indicates a greater likelihood of prescribing concomitant pathogenetic therapy in comparison with traditional treatment. In addition, using a representative group (n = 3431), it was found that when using mebeverine, the likelihood of hospitalization for various diseases increases, which indicates the variability of the therapeutic effect of the drug and is accompanied by an increase in treatment costs (Table 3).
IN
The main indications for prescribing antispasmodics for diseases and conditions of internal organs have been formulated.
Conclusion
Pain is a universal symptom for a wide variety of lesions of internal organs; its occurrence is often based on spasm of smooth muscles. Knowledge of the clinical pharmacology of drugs in this group allows for the most targeted choice of drug. Using the pharmacodynamics of drugs, it becomes possible to combine them rationally, effectively and safely. Of the entire group of antispasmodic drugs, the most popular is drotaverine (No-spa), which meets the criteria of effectiveness, safety and cost-effectiveness to a greater extent than the others. A review of research results shows that No-Spa is effective both for the rapid relief of acute pain caused by spasms, and for a fairly long-term treatment of patients with chronic pathology. Unlike other antispasmodics, the effect of No-shpa is not limited to the gastrointestinal tract, which makes the drug universal for relieving spasmodic conditions of any origin and location and the associated pain syndrome. An important feature of the pharmacodynamics of No-shpa is the lack of effect on cholinergic structures, which makes the drug safer compared to M-anticholinergics. The mechanism of action, which is different from other drugs in the group, provides opportunities for a wide effective combination both within the group of antispasmodics and with drugs from other pharmacological groups (antisecretory, non-narcotic analgesics, opioids).
For questions regarding literature, please contact the editor.
M. V. Pchelintsev , Candidate of Medical Sciences, Associate Professor Institute of Pharmacology of St. Petersburg State Medical University named after. acad. I. P. Pavlova , St. Petersburg
Phosphodiesterase inhibitors (PDE) type IV
These are traditionally widely used antispasmodics: drotaverine hydrochloride and papaverine. Their injectable forms are sold with a prescription, while their tablet forms are available without a prescription. PDE IV is widely present in smooth muscle along the entire length of the intestine, as well as in the biliary and urinary tracts. Thus, blocking PDE IV has a universal antispasmodic effect regardless of the severity of the spasm or its cause. In addition, drotaverine has anti-edematous and anti-inflammatory effects. Drotaverine can be used for acute abdominal pain - both to relieve acute spasms and for long-term treatment of biliary dyskinesia, cholelithiasis, chronic intestinal diseases with spastic syndrome, which are accompanied by colicky or bursting pain. Unlike M-anticholinergics, drotaverine can be used in elderly men with prostate pathology, as well as with concomitant pathology and combined use with other drugs. The therapeutic concentration of drotaverine in plasma when taken orally is achieved after 45 minutes.
Research results and discussion
Initially, on days 3–5 (removal of the urethral catheter), the indicators in the groups were not statistically significantly different. According to the questionnaires, all patients had irritative symptoms that reduced the quality of life. Comparative dynamics of the study results in the observation groups are presented in Table 1.
After 1 month. therapy, positive dynamics were noted in both groups. Statistically significant (p<0.05) in comparison with the initial results, the frequency of urination and nocturia decreased, Qmax increased, and the average score on the IPSS and QOL scales increased. However, the values of these indicators in group 2, in turn, differed significantly (p<0.05) from those obtained in group 1 and were closer to normal.
In group 1, no adverse events were noted. The proportion of patients with side effects in group 2 was 16.7%, but there were no cases of refusal to further participate in the study. Adverse events were identified as dry mouth (13.3%) and constipation (3.3%).
By the end of the 3rd month. observation in group 1, no statistically significant dynamics of parameters were revealed (p>0.05) (Table 1). In group 2, while taking fesoterodine, there was a further significant (p<0.05) decrease in the number of urgent urges, a decrease in the number of daytime and night urination, as well as an increase in Qmax, a decrease in the average score on the IPSS scale and an improvement in the quality of life of patients according to the scale QOL. In this group, these indicators reached normal values.
It should also be noted that there were no new cases of adverse events and episodes of acute urinary retention in both the 1st and 2nd groups.
At the end of 6 months. therapy in both groups, no significant changes were recorded compared to the results of the previous visit, and no new cases of adverse events and acute urinary retention were identified.
As evidenced by the results obtained during the study, all patients with BPH who underwent TUR of the prostate had symptoms of filling after removal of the urethral catheter. This may be explained by both inflammation in the lower urinary tract that persists in the postoperative period and detrusor overactivity. Both groups received anti-inflammatory therapy to eliminate the inflammatory component.
Taking fesoterodine in the 2nd group of patients made it possible to achieve significantly (p<0.05) more significant positive results by the 30th day, which manifested itself in more effective relief of dysuria, an increase in Qmax due to an increase in the effective capacity of the bladder and contributed to improved quality patients’ lives, according to the QOL scale, as evidenced by the preliminary results of the study [12].
Further administration of fesoterodine for 3 months. allowed us to completely normalize the studied parameters in group 2 (against the background of the absence of significant dynamics in group 1), which corresponds to the results of previously published large studies [13–16].
With regard to the safety of therapy with m-anticholinergics in this category of patients, it should be noted that, despite the relatively larger number of adverse events in the group of patients receiving fesoterodine, they were mild or moderate in nature (dry mouth, constipation), none of them led to to patient dropout from the study. However, no serious adverse events or cases of acute urinary retention were recorded. Thus, fesoterodine has a relatively high safety profile among drugs in this group, which is also consistent with the results of studies conducted in European countries [17, 18].
Sodium channel blockers
These drugs block intracellular calcium stores through a cascade of reactions. The most famous drug is a derivative of veratric acid - mebeverine. Unlike anticholinergics, it does not cause hypotension of the colon. Most often, this drug is prescribed for functional disorders of the gastrointestinal tract or as an adjuvant for organic diseases of the gastrointestinal tract. Mebeverine is used only for course treatment. Mebeverine is most effective in combination with other pathogenetic drugs. Mebeverine is mainly available by prescription, but there is a dosage form in tablets that does not require a doctor's prescription.
We will not dwell on calcium channel blockers in more detail, since these drugs do not have any over-the-counter forms.
The over-the-counter myotropic antispasmodic trimebutine stands apart. Trimebutine acts on the enkephalinergic system of the intestine and peripheral opiate receptors - µ, κ and δ. Having an affinity for receptors that enhance and suppress peristalsis, it stimulates the contraction of intestinal smooth muscles in hypokinetic conditions and is an antispasmodic in hyperkinetic conditions. Trimebutine reduces the tone of the esophageal sphincter, promotes gastric emptying and increased intestinal motility, as well as the response of the smooth muscles of the colon to food irritants. It is used for various functional disorders of the gastrointestinal tract.
Read also[edit | edit code]
- Anatomy and physiology of the nervous system
- Parasympathetic nervous system
- Sympathetic nervous system
- Synaptic transmission
- Acetylcholine
- Cholinergic receptors and synapses
- Cholinomimetics
- Nicotine
- M-cholinergic receptor stimulants
- M-cholinergic receptor blockers
- Acetylcholinesterase inhibitors (AChE)
- Poisoning with acetylcholinesterase blockers
- Nerve transmission at neuromuscular synapses and autonomic ganglia N-cholinergic receptors
- Muscle relaxants
- Agents acting on the autonomic ganglia
- Ganglion stimulators
- Ganglioblockers
Ask and choose
How can a pharmacy client choose an over-the-counter antispasmodic from the extensive list of drugs for abdominal pain? Guiding questions will help:
- Does a person have a chronic organic gastrointestinal disease - peptic ulcer of the stomach or duodenum, chronic cholecystitis, chronic pancreatitis, biliary colic, biliary dyskinesia? As a rule, the patient already knows what helps him or what drug the doctor recommended for him during an exacerbation.
- Patient's age? Selective M-anticholinergics are not suitable for the elderly due to the risk of developing intestinal obstruction. Also, antispasmodics have age restrictions for children: mebeverine can be used from twelve years of age, hyoscine butylbromide and drotaverine - from six, trimebutine - from three.
- For women - pregnancy, lactation? The use of antispasmodics during pregnancy is possible only as prescribed by a doctor. PDE inhibitors and trimebutine are contraindicated during lactation. Anticholinergics and sodium channel blockers can be used during lactation.
- Pain in the upper abdomen, possibly accompanied by nausea or vomiting? A selective M-anticholinergic drug is suitable for adults, in the absence of contraindications described in the instructions for a particular drug.
- Pain in the lower abdomen? Type IV PDE inhibitors and sodium channel blockers are suitable.
Sources
- Abdominal pain syndrome in the practice of general practitioner T. M. Benz “News of medicine and pharmacy.” Gastroenterology (239), 2008.
- Pchelintsev M. V. “Anspasmodics: from clinical pharmacology to pharmacotherapy.” Attending Physician 7 (2008): 74–77.
- Mikhailov I. B. Clinical pharmacology (textbook for students of medical universities) - 5th ed., revised. and additional - St. Petersburg: publishing house "Sotis-Med", 2013. - 588 p.
- Yakovenko E. P., et al. "Opiate receptor agonist trimebutine in the treatment of functional disorders of the gallbladder and sphincter of Oddi." Journal "Attending Physician" 2–2014 (2017): 56.
- Tytgat GN. Hyoscine butylbromide: a review of its use in the treatment of abdominal cramping and pain. Drugs 2007; 67:1343–57.