1.What is sickle cell anemia and its causes
Sickle cell anemia is an inherited blood disorder.
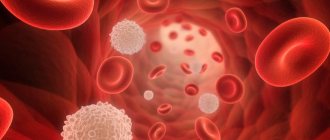
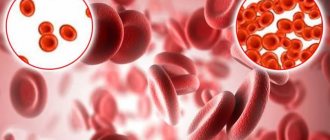
Let's try to figure out what it is. Red blood cells contain hemoglobin, a protein that carries oxygen in the blood. Normally, red blood cells are round in shape and flexible, allowing them to move through small blood vessels to deliver oxygen to all parts of the body.
Sickle cell anemia causes blood cells to take the shape of a crescent, or sickle. In this form of blood cells, red blood cells break down very easily, which causes anemia
. Sickle red cells live only 10-20 days instead of 120 days, as is normal for healthy cells. In addition, damaged red blood cells clump and adhere to the walls of blood vessels, blocking blood flow. This can cause severe pain and permanent damage to the brain, heart, lungs, kidneys, liver, bones and spleen.
Causes of sickle cell anemia
Human sickle cell anemia is caused by a genetic abnormality in the hemoglobin gene. This abnormality results in sickle hemoglobin. When oxygen is released from sickle hemoglobin, it sticks together and forms long rods that damage the red blood cell and change its shape. Sickle-shaped red blood cells cause the symptoms of sickle cell anemia.
Human sickle cell anemia is not contagious. This is a genetic disease that a person is born with. The disease develops when a child inherits two abnormal hemoglobin genes, one from each parent. People who have inherited only one abnormal hemoglobin gene are carriers of it, but they will not have anemia or symptoms of the disease.
A must read! Help with treatment and hospitalization!
Publications in the media
Sickle cell anemia is the most commonly reported hereditary hemoglobinopathy, characterized by moderate chronic hemolytic anemia, recurrent acute painful crises and increased susceptibility to infectious diseases (mainly Streptococcus pneumoniae) due to the formation of pathological HbS. The disease is diagnosed in 1% of African Americans and is often observed in Azerbaijan and Dagestan.
Etiology • At the molecular level. HBB gene defect (*141900, 11p15.5, classic mutation: HbS is formed as a result of the replacement of valine with glutamic acid in position 6 of the b-chain of the Hb molecule, also other mutations, Â). In the venous bed, HbS polymerizes to form long chains that change the shape of red blood cells (become sickle-shaped) • At the cellular level. Sickle-shaped red blood cells cause an increase in blood viscosity, stasis; create a mechanical barrier in small arterioles and capillaries, leading to tissue ischemia (which is associated with pain crises). In addition, sickle erythrocytes are less resistant to mechanical stress, which leads to their hemolysis.
Clinical picture: moderate jaundice, trophic ulcers in the ankle area, retardation in physical development (especially in boys), priapism, predisposition to aplastic and hemolytic crises, pain crises, splenomegaly, cholelithiasis, avascular necrosis, leg ulcers, osteonecrosis with the development of osteomyelitis.
Diagnostics • The diagnosis is confirmed by the detection of polymerized HbS during Hb electrophoresis • Other criteria: •• anemia - Hb about 80 g/l; mean erythrocyte volume >75 µm3 •• reticulocytosis (hyperregenerative anemia) •• leukocytosis •• thrombocytosis •• peripheral blood smear: sickle-shaped red cells, polychromasia •• serum bilirubin concentration slightly elevated (20–40 mg/l [34–68] µmol/l]) •• ESR is reduced •• serum LDH content is increased •• haptoglobin is absent •• in the bone marrow - erythroid hyperplasia, an increase in multinucleated precursor cells.
Treatment • For acute pain crises of moderate severity in an outpatient setting - non-narcotic analgesics (ibuprofen, paracetamol) • For acute pain crises in an inpatient setting - narcotic analgesics parenterally. You should reduce the dose of the narcotic analgesic as quickly as possible, and then switch to taking non-narcotic analgesics, for example paracetamol, ibuprofen • The possibility of dehydration and acidosis must be taken into account • If an infection develops, before obtaining the results of a bacteriological study, an antibiotic active against Streptococcus pneumoniae and Haemophilus influenzae should be prescribed • Maintenance therapy - transfusions of washed or thawed red blood cells, as well as anticoagulants • Bone marrow transplantation.
Complications • Aseptic necrosis of the femoral head and other necrotic complications • Sepsis • Cerebrovascular disorders with neurological manifestations • Cardiomegaly • Retinopathy • Hemosiderosis.
Course and prognosis. Anemia is radically incurable. In the second decade of life, the number of crises decreases, but complications arise more frequently. Some patients die in childhood from necrotic complications or sepsis. Most patients live to an average age of 50 years.
Synonyms • Drepanocytosis • Meniscocytosis • Sickle cell syndrome • Meniscocellular anemia • Meniscocytic anemia
ICD-10 • D57 Sickle cell disorders
2. Symptoms of the disease
Symptoms of sickle cell anemia may include:
- Severe pain;
- Anemia;
- Chest pain and difficulty breathing;
- Joint pain, arthritis;
- Blockage of blood flow in the spleen or liver;
- Severe infections.
People with sickle cell disease develop severe pain in the chest, back, arms, legs, and abdomen. Additionally, pain can occur in any part of the body. Sickled red blood cells in the lungs can cause serious illness with symptoms such as chest pain, fever and difficulty breathing. Sickle cell anemia can also cause permanent damage to the brain, heart, kidneys, liver, spleen and bones. The special thing about this disease is that the severity and symptoms of the disease can vary greatly from person to person, even if they are blood relatives.
Visit our Therapy page
Types
There are several different types of sickle cell disease. The main types include:
- HbSS
- Sickle cell disease (homozygosity for abnormal HbS): A person inherits two genes, one from each parent, for sickle cell disease. He will have sickle cell anemia, which is the most severe type of sickle cell anemia. - HbSC is a double heterozygous (compound heterozygous) carrier of two abnormal hemoglobins HbS and HbC
: a person inherits the sickle cell gene from one of the parents. A gene is inherited from the other parent that results in a different type of abnormal hemoglobin. This disease is usually less serious. - Sickle cell beta thalassemia HbS
: A person inherits the sickle cell gene from one parent and the gene for beta thalassemia, another type of anemia, from the other parent. - Carriage of the sickle cell trait
: If a person has only one gene, they will not have sickle cell disease, but they can pass the gene on to their children.
Sickle cell disease can affect anyone, but is more common in black people. Sickle cell anemia is more likely in areas where malaria is common. However, patients with sickle cell disease have a lower risk of some severe types of malaria.
3. Diagnosis of sickle cell anemia
Sickle red blood cells can be seen when a blood sample is examined under a microscope. But to diagnose human sickle cell anemia, a special blood test is used - hemoglobin electrophoresis
, which measures the amount of abnormal hemoglobin. Depending on the amount of sickle hemoglobin measured, it can be determined whether a person is a carrier of the gene or has sickle cell disease.
There are also quick screening tests,
which help identify sickled red blood cells or clumps of abnormal hemoglobin when oxygen leaves the blood. But these tests are rarely used because they cannot distinguish between disease and the simple presence of a disease gene.
Perinatal diagnosis of sickle cell anemia
carried out by studying the DNA of fetal cells obtained through chorionic villus sampling or amniocentesis.
About our clinic Chistye Prudy metro station Medintercom page!
Sickle cell anemia
Terminology
For more information about the composition and biological role of blood, about the essence of anemia, see the material “Anemia.
Blood and bloodlessness." Sickle cell anemia is a severe hereditary disease, also known as drepanocytosis, meniscocytosis, S-hemoglobin disease, Gerrick's syndrome, African hemolytic anemia (hemolytic - Greek lit. "dissolving, decomposing blood"). The modern history of the study of this type of anemia begins, it is believed, in the middle of the 19th century , when an autopsy revealed no spleen in the body of an African slave executed for escaping. Then in 1910 a report appeared, authored by cardiology professor James B. Herrick and his intern Ernest E. Irons. In a blood test of a 20-year-old student who had been treated since 1904 for “anemia, muscular rheumatism and biliousness” (he died of pneumonia in 1916), Irons observed and first described red blood cells of a strange shape, which he defined as “elongated and sickle-shaped.” " The name "sickle cell anemia" was first used as a diagnosis by Verne Mason in 1922 . Since the 1930s, epidemiological studies of this disease began in samples of children of African descent.
It can be assumed that the genetic mutation leading to sickle cell anemia appeared on the African continent - and, oddly enough, became established in the population as a useful trait: carriers of the gene and patients with this form of anemia show relative resistance to malaria. The malarial plasmodium parasitizes red blood cells and destroys them, however, red blood cells, genetically mutilated and carrying a completely different hemoglobin (see below), are “inedible” for plasmodium. And although the anemia of malaria is not sweeter, the mortality rate of swamp fever is still higher: those who had at least some immunity survived.
In an actively migrating humanity, which is becoming increasingly international and gradually interracial, sickle cell anemia is widespread. However, a certain endemicity remains today: in Equatorial Africa, the Mediterranean basin, India, and the Middle East, the frequency of genetic carriage (not to be confused with the incidence in the clinical form) reaches 40%. According to a special study conducted in 2001 in Jamaica, the average life expectancy of patients with homozygous sickle cell anemia is 53 years for men and 58 years for women. About 90% live to be 20 years old, and about half live to be 50 years old.
In other words, the problem is very serious, it is characterized by high medical and social significance and requires effective solutions, the search for which is currently being carried out by leading specialized research centers. The prevalence of hereditary hemoglobinopathies (including sickle cell anemia) in the world is estimated at 3-7%. In the early 2010s, WHO classified sickle cell disease as a global health problem.
The epidemiological situation and mortality rates in third world countries are unknown or not sufficiently reliable.
According to the literature, due to migration processes, the incidence in Russia shows a significant and stable tendency to increase, especially in Moscow and St. Petersburg.
Lifestyle
Many people with sickle cell disease can live full and active lives, especially if they take steps to reduce the effects of the condition. Lifestyle measures that may help include:
- regular medical examinations
- drink 8 to 10 glasses of water per day
- do not overcool or overheat, as this can provoke a crisis
- engage in physical activity, but also get enough rest
- maintaining a healthy diet.
Scientific article on the topic: Gene therapy will help cure sickle cell anemia.
Sickle cell anemia - treatment
Drug treatment
The following medications may reduce the risk of complications:
- Hydroxyurea
(Hydrea): Helps provide oxygen to the body. It is not safe to use during pregnancy. Studies have not proven the benefit of this drug in children younger than 9 months. - L-Glutamine Oral Powder
(Endary): Helps reduce the number of sickle cells and is suitable from age 5. - Voxelomotor
(Oxbrita): Increases healthy hemoglobin levels. Suitable from 12 years of age. - Crizanlizumab-tmca
: Reduces pain by preventing blood cells from attaching to blood vessels. Suitable from 16 years of age.
Prevention of infections
Doctors recommend that adults and children take precautions to reduce the risk of infection. These include:
- regular hand washing
- stay away from reptiles such as turtles as they can carry salmonella
- Get vaccinations against influenza, pneumococcal disease, and meningococcal disease
Your doctor may prescribe penicillin or another antibiotic for children under 5 years of age.
Blood transfusion and stem cell transplantation
People with severe symptoms may require a blood transfusion or stem cell transplant. Blood transfusions may be necessary if the patient has severe anemia, an enlarged spleen, infection, or other complications.
A stem cell transplant using cells from a healthy donor can cure sickle cell disease, but can be risky. In addition, the stem cells must be precisely selected.