Порфирии — группа генетически обусловленных патологий, проявляющихся на фоне нарушения синтеза гема (промежуточного продукта метаболизма гемоглобина) и накопления его токсичных соединений в организме человека. Симптоматика заболеваний разнообразна: пациенты могут сталкиваться со светочувствительностью, высыпаниями на коже, хроническими болями в животе, частичным или полным параличом, острыми психозами. Диагностика метаболического расстройства осуществляется посредством молекулярно-генетических тестов и лабораторных анализов биоматериалов, полученных врачами от ребенка или взрослого. Медикаментозная терапия направлена на снижение концентрации токсических метаболитов в крови пациента. Хирургические вмешательства выполняются при осложненном течении патологии.
Общие сведения
Болезнь порфирия (син. порфириновая болезнь) представляет собой большую группу заболеваний обмена веществ, обусловленных преимущественно наследственным дефектом в системе биосинтеза гема (соединение ионов железа с производными порфирина) и накоплением в организме его токсичных метаболитов (порфобилиногена/δ-аминолевулиновой кислоты). Большинство порфирий являются врождёнными заболеваниями с наследованием по аутосомно-доминантному типу. Значительно реже порфирии, обусловленные нарушением метаболизма, являются приобретенным и развиваются под воздействием различных факторов, способствующих процессу ингибирования ферментов синтеза гема.
В человеческом организме происходит постоянный эндогенный синтез пуринов/пиримидинов. К пуринам относятся ксантин, аденин, гипоксантин, гуанин; к пиримидинам – цитозин, урацил, тимин, оротовая кислота. Они необходимы для хранения, транскрипции/трансляции генетической информации, деления/роста клеток, передачи сигналов и накопления энергии.
Порфирины в человеческом организме синтезируются в клетках костного мозга, печени, тканях нервной системы, поджелудочной железе и находятся как в свободном, так и связанном состоянии, образуя различные сложные белковые соединения с ионами железа (гемоглобин, миоглобин, цитохром, пероксидазу, хромопротеид) или комплексы с натрием, калием, медью, ванадием, никелем, оловом, цинком, марганцем, кобальтом. При этом, механизм синтеза порфиринов одинаков в клетках всех тканей, однако скорость их образования/длительность существования существенно варьирует. Основной функцией порфиритовых комплексов является их участие в сложных метаболических процессах (транспортировка кислорода, биологическое окисление, фотосинтез и др.). Конечным продуктом пуринового метаболизма является мочевая кислота. Порфирины выделяются из организма с мочой, калом и желчью.
Практически любая нозологическая форма порфирии реализуется вследствие снижения активности ферментов в цикле биосинтеза гема (торможение процесса синтеза гема), что обусловлено мутацией в патогномоничном гене и приводит к накоплению промежуточных токсичных метаболитов. Однако это генетическое заболевание не всегда манифестирует выраженной симптоматикой, поскольку патология не всегда себя проявляет даже при снижении активности фермента до 50% от нормы. И только 18-20% генетических носителей имеют характерную клиническую симптоматику.
В целом порфириновая болезнь относятся к достаточно редким заболеваниям: обобщенный показатель заболеваемости различными формами болезни составляет 1:20 000 (Википедия). При этом, показатели заболеваемости на 100 тыс. населения широко варьирует — наследственная копропорфирия: 3–5 случаев; перемежающаяся острая перемежающаяся порфирия: в пределах 5–10 случаев; вариантная порфирия – 2–3 случая; поздняя кожная порфирия – 15–20 случаев на 100 тыс. населения. Порфирии не эндемичные заболевания (то есть характерные для определенной местности) и встречаются среди населения различных стран с одинаковой частотой.
Заболевание порфирия протекает как хронически, так и в виде острых атак. Существенно варьирует и возраст дебюта заболевания: эритропоэтические порфирии манифестируют преимущественно в 3-5 лет (дошкольном детстве), острые порфирии развиваются в 14-16 лет (период/после полового созревания), приобретенная спорадическая форма печеночной кожной порфирии — у лиц после 40 лет. Ввиду обширности темы рассмотрим лишь некоторые формы порфирий, в частности болезнь Гюнтера.
Патогенез
Патогенез всех форм порфирий имеет общие звенья, независимо от особенностей клинического течения/тканевой принадлежности. В их основе — отсутствие/снижение активности определённого фермента в общей цепи биосинтеза гема, что приводит к избыточному накоплению продукции в токсических концентрациях порфиринового обмена перед звеном, где локализуется дефектный энзим. Локализуются гены ферментов на разных хромосомах и групповой сцепленности не имеют.
Процесс синтезирование гема включает восемь последовательных этапов и каждый фермент отвечает свой этап и кодируется определенным геном. Соответственно каждая форма порфирии имеет свой специфичный ферментативный дефект. При хронических формах порфирий отмечается накопление копропорфирина, протопорфирина и уропорфирина; при острых формах — порфобилиногена/ДАЛК (дельта-аминолевулиновой кислоты).
Классификация
В основу квалификации порфирий положены такие признаки как место нарушения метаболизма порфиринов/накопления порфиринов и клинические проявления, согласно которым выделяют:
- по месту нарушения метаболизма порфиринов различают: эритропоэтические (первичное нарушение в костном мозге) и печеночные порфирии (нарушения развиваются первично в печени);
- по клиническим проявлениям порфирии делятся на острые формы, проявляющиеся преимущественным поражением нервной системы и формы, манифестирующие поражением кожи.
Соответственно, каждая из форм включает несколько видов порфирий. Обобщенная классификация форм и видов порфирий приведена в таблице ниже.
Диагностика порфирии
Относительная редкость патологии и разнообразие клинических проявлений становятся причиной регулярных диагностических ошибок. Особенно это касается диагностики печеночных порфирий. Часто такие пациенты безрезультатно лечатся у дерматолога и других узких специалистов.
Для диагностики заболевания необходимо провести анализ на определение уровня различных фракций порфиринов в крови, моче и кале. В зависимости от того, уровень каких фракций порфиринов повышен и каково соотношение между разными фракциями, диагностируется та или иная форма порфирии.
Причины
Причиной порфирий в подавляющем большинстве случаев являются мутации в генах, способствующие снижению активности фермента, принимающего участие в процессе биосинтеза гема. Наследуется заболевание преимущественно по аутосомно-доминантному, реже по аутосомно-рецессивному типу (болезнь Гюнтера). И только поздняя кожная порфирия (урокопропорфирия) может развиваться как вследствие длительной интоксикации тяжелыми металлами/заболеваний печени (опухоли печени, алкогольный/вирусный гепатит С), так и развивается при наличии наследственной предрасположенности.
Однако, для развития некоторых форм заболевания, в частности острая порфирия, кроме присутствия патологической мутации в гене фермента для манифестации симптоматики требуется воздействие провоцирующих факторов, которые стимулируют процесс выработки порфиринов. К такими общепризнанным факторам относятся сильные/постоянные стрессы, длительная инсоляция, злоупотребление алкоголем, голодание, инфекции бактериального/вирусного генеза, интоксикация тяжелыми металлами (ртуть, свинец), лекарственные средства (седативные средства, НПВС, сульфаниламиды, оральные контрацептивы, цефалоспорины, антималярийные препараты, барбитураты и др.), у женщин — менструальный цикл/беременность.
Наличие этих факторов способствует или повышенному потреблению гема, как конечного продукта или стимуляции активности начального фермента цикла биосинтеза. Как следствие — ускоряется синтез/накопление промежуточных продуктов обмена порфиринов в токсических концентрациях.
Терапевтический курс
Лечение острой и эритропоэтической порфирии требует госпитализации пациента. Лица, страдающие ПКП, могут проходить терапию в амбулаторных условиях. Современная медицина не располагает средствами, позволяющими полностью устранить нарушения в обмене порфиринов. Усилия врачей направлены на устранение симптомов заболевания и факторов, провоцирующих его обострения.
При острых формах патологии ребенку или взрослому назначаются производные аденозинтрифосфата, подавляющие синтез токсичного порфобилиногена. Дальнейшее лечение предусматривает прием пациентом больших дох глюкозы и переход на высокоуглеводную диету.
Эритропоэтический тип заболевания почти не поддается лечению. Основной терапевтической мерой становится ограничение пребывания пациента под солнечными лучами. Эрозии следует обрабатывать антисептическими средствами для предупреждения инфекционных поражений кожных покровов. Выраженный гемолиз (разрешение эритроцитов) становится показанием к удалению селезенки. В отдельных случаях пациентам показана трансплантация костного мозга.
При поздней кожной порфирии мужчинам и женщинам назначается плазмаферез и медикаментозная терапия. Применяемые препараты призваны снизить уровень всасывания порфиринов в ЖКТ.
Симптомы
Клинические проявления порфирий варьируют в широких пределах и определяются конкретной формой заболевания. Рассмотрим лишь несколько форм заболевания.
Болезнь Гюнтера (врожденная эритропоэтическая порфирия)
Как и все другие эритропоэтические порфирии болезнь Гюнтера достаточно редкое явление. Манифестирует заболевание уже в раннем детстве (в возрасте 1-5 лет) и обусловлено врожденными ферментопатиями. Местом нарушения порфиринового обмена являются эритробласты костного мозга. Регистрируется в семьях исключительно среди сестер/братьев одного поколения и наследуется от любого из родителей как аутосомно-рецессивное. При этом, у родителей больных детей какие-либо клинические/биохимические признаки болезни отсутствуют. Для больных эритропоэтической порфирией характерен дефицит косинтетазы уропорфириногена, который концентрируется в патологической популяции эритробластов и в результате такой ферментативной блокады нарушается биосинтез гема, что приводит к накоплению в организме ребёнка критической концентрации уропорфирина I. Встречаются одинаково часто у лиц мужского/женского пола.
Для болезни Гюнтера характерны шесть основных признаков:
- Повышенная чувствительность к солнечной радиации, свету с появлением на открытых участках кожи пузырей.
- Отсутствие типичной симптоматики для острой порфирии (абдоминальных симптомов).
- Чрезмерное развитие волосяного покрова.
- Выделение окрашенной в красный цвет мочи.
- Розовато-коричневая окраска зубов.
- Увеличение селезенки на фоне гемолитической анемии.
Манифестировать заболевание может как всеми признаками, так отдельными из них. Изменения со стороны кожи возникают преимущественно ранней весной на фоне сниженного диуреза и общей слабости. Проявляются изменения зудом/покраснением кожи на тыльной стороне кистей/стоп, ушных раковинах, лице, на голени и предплечьях после инсоляции, на месте которых позже появляются буллезные элементы (пузырьки) с серозно-геморрагическим содержимым. При этом кожа чрезвычайно чувствительна к механическим воздействиям. В случаях присоединении вторичной инфекции происходит изъязвление пузырьков и заживление с образованием рубцов.
Кожа неровная, кисти имеют когтеобразный вид, ногтевые пластинки утолщены/деформированы/помутневшие; в ряде случаев при хроническом течении разрушаются ушные раковины, отмечается мутиляция фаланг кисти с нарушением ее функции. На рентгенограмме остеопороз эпифизов фаланг, контрактура суставов (частичная/полная) в дистальных отделах конечностей. Для пациентов характерны длинные ресницы/густые брови, слабое физическое развитие, общее истощение, гиперпигментация, бледность кожи, окрашенные зубы, гипертрихоз на лице, увеличенная селезенка. Реже — помутнение хрусталика/роговицы. Лабораторно в кале/моче, плазме и в эритроцитах отмечается высокое содержание уро/копро/протопорфирина.
Поздняя кожная порфирия
Наиболее часто встречаемая порфирия печени, обусловленная нарушением процесса синтеза гемов печени, при которой отмечается повышенное образование/выделение копропорфирина/уропорфирина с мочой и их задержкой в кожных покровах. Урокопропорфирия манифестирует тремя основными дерматологическими симптомами: пигментация; пузыри; гипертрихоз.
Гиперпигментация кожи носит диффузный характер и возникает преимущественно на участках тела, подвергающихся солнечной инсоляции (кисти рук, шея, лицо, ушные раковины, верхняя часть груди). Цвет варьирует от красновато-синюшного (бронзового) до землисто-серого и определяется индивидуальными особенностями, наличием сопутствующих заболеваний и непосредственным влиянием внешних климатических/профессиональных факторов. Такая характерная реакция возникает лишь на местах облучения, и она расценивают как фотосенсибилизация (фотодерматоз), который при отсутствии облучения устраняется. Реакция усиливается летом, а зимой практически исчезает. По прошествии времени пигментация приобретает стойкий характер и становится более интенсивной, слабо изменяясь на протяжении года.
Следующим классическим признаком является пузырная реакция, которой предшествует повышенная ранимость кожи тыльной стороны кистей/лица. Проявляется образованием на видимо неизмененной коже пузырей размерами от просяного зерна до горошины округлой/овальной формы с жидкостным содержимым (вначале серозным, а затем мутнеет и переходит в гнойное). В дальнейшем пузыри самопроизвольно вскрываются, образуя эрозии с неправильными очертаниями с формированием впоследствии атрофических рубцов. Пузырная реакция увеличивается в весенне-летний период, а зимой пузыри, как правило, отсутствуют. Преимущественной локализацией пузырей являются кожа лица, шеи, ушных раковин, тыла кистей; реже — на волосистой части головы, предплечьях, губах. У женщин может иметь место атипичное расположение пузырей — на закрытых участках кожи: бедрах, спине, голенях. Длительность пузырной реакции варьирует от 1-2 недель до нескольких месяцев. В случаях присоединения вторичной инфекции отмечается лимфаденит/лимфангоит, глубокие язвы. Гипертрихоз – менее специфический синдром, встречается в среднем у 70% пациентов на лице в лобно-височной области.
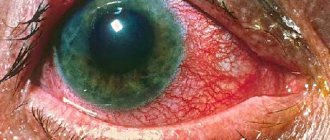
Для этой формы свойственны ожирение, нарушение функций ЖКТ, поражения печени, органа зрения: нарушение цветовосприятия, помутнение роговицы, конъюнктивит, расширение сосудов глазного дна. Кроме этих основных симптомов характерно преждевременное старение кожи и появление глубоких морщины. Ногти также подвергаются изменению: деформируются, теряют блеск, становятся матовыми; часто развивается подногтевой гиперкератоз. Пациенты выглядят старше своего возраста. Ниже приведены фото больных порфирией (поздняя кожная форма).
При острых порфириях развиваются сильные боли в животе, задержка стула, учащение сердцебиения, повышение артериального давления, изменение цвета мочи (от розового до красно-бурого). Тяжесть состояния пациента в основном обусловлена неврологическими симптомами – болью по всему телу, снижением чувствительности, прогрессирующей мышечной слабостью, иногда достигающей полного паралича, судорожными припадками, различными психическими расстройствами (тревожность, психомоторное возбуждение, бред, галлюцинации).
Острые порфирии: острая перемежающаяся порфирия
Встречается значительно чаще у лиц женского пола. В развитии заболевания существенную роль играют провоцирующие факторы. Для этой формы заболевания характерны несколько групп симптомов:
- Болевой синдром — интенсивные боли в животе, сопровождаемые тошнотой/рвотой, расстройствами стула (диарея/запоры); боли носят приступообразный характер, реже – постоянный и могут продолжаться от 5-6 часов до нескольких дней с локализацией в различных отделах живота.
- Неврологическая симптоматика манифестирует вялыми параличами; парезами; полиневритом; сенсорными/бульбарными нарушениями, расстройством функции уретрального сфинктера.
- Психические расстройства (эмоциональная лабильность, хроническая бессонница, депрессии/тревоги, психомоторное возбуждение, бред, слуховые/зрительные галлюцинации, делирий).
- Гипоталамическая дисфункция (лихорадка центрального генеза, гипонатриемии).
- Эпилептиформные припадки с высоким риском развития во время острых приступов коматозного состояния.
- Нарушения со стороны сердечно-сосудистой системы — гипертензия, синусовая тахикардия, изменения на ЭКГ.
- Пигментурия (розовая/красная моча, темная диффузная окраска кожи, хлоазмы, веснушки).
Клиническая симптоматика и ее выраженность определяется формой заболевания. Так, при скрытом/здоровом носительстве мутантного аллеля клинические признаки отсутствуют на фоне присутствия минимальных биохимических отклонений от нормы.
- Для латентной формы характерны мышечная слабость, периодически возникающие боли в животе, синусовая тахикардия, бессонница, гипертензия, реже — психологические изменения личности.
- Манифестные острая форма перемежающейся порфирии может протекать в нескольких вариантах. Лёгкая форма характеризуется непродолжительными периодически возникающими острыми приступами болезни, ограничивающиеся абдоминальными симптомами, которые заканчиваются в большинстве случаев благоприятно. Тяжёлая форма: характерны тяжёлые приступы длительностью от 2 до 10 недель с рецидивами через несколько месяцев/1-2 года. Манифестирует абдоминальными симптомами, психическими расстройствами/неврологическими нарушениями. Может закончиться летально. Ступенчатая форма: для нее характерным является нарастание симптоматики и глубоких общих нарушений с каждым новым приступом. Рецидивы учащаются и наступают через 1-2 месяца. В большинстве случаев исход неблагоприятный и каждый 3-4 приступ заканчивается летально. Острейшая форма: крайне тяжелое течение, сопровождаемое тяжелыми общими нарушениями; чаще встречается во время беременности и 24-60% случаев заканчивается летально от паралича дыхательного центра.
Клинический случай
Пациент Т
., 20 лет, 23.12.17 во время тренировки по спортивным единоборствам получил удар в живот, после чего отмечал эпизод тошноты. Через 4 дня появились выраженная боль в эпигастральной области, тошнота, рвота желчью. Лечение не получал, в течение 2 дней симптоматика регрессировала. 05.01.18 вновь почувствовал боль в эпигастральной области, сопровождавшуюся рвотой и жидким стулом, температура тела при этом не повышалась. Через 2 сут госпитализирован в районную больницу по месту жительства с диагнозом «острый панкреатит». Перенесенные ранее заболевания и обращения за медицинской помощью отрицал. Назначена антисекреторная терапия, рекомендован голод. За 2 нед потерял массу тела на 13 кг, появились слабость и боли в конечностях. 16.01.18 г. пациент переведен в отделение реанимации и интенсивной терапии (ОРИТ) областной больницы. При обследовании обнаружены электролитные нарушения (уровень натрия — 128 ммоль/л, калия — 2,8 ммоль/л), амилаземия (уровень амилазы — 223 МЕ/л), стеноз устья чревного ствола (80% по данным компьютерной томографической ангиографии). Консервативная терапия к улучшению состояния пациента не привела, в связи с чем 28.01.18 он госпитализирован в Федеральный научно-клинический центр специализированных видов медицинской помощи и медицинских технологий с диагнозом «Стеноз чревного ствола. Синдром хронической абдоминальной ишемии. Кахексия. Электролитные нарушения» для решения вопроса об оперативном лечении. При поступлении: состояние пациента тяжелое. Кожные покровы обычной окраски, нормальной влажности. Температура тела 37,7 °С. Дыхание самостоятельное, везикулярное, с частотой 18 в 1 минуту, насыщение гемоглобина крови кислородом 98%. Артериальное давление — 155/110 мм рт.ст., частота сердечных сокращений — 115 уд/мин. По данным электрокардиографии — синусовая тахикардия. Живот не вздут, при пальпации мягкий, безболезненный, симптомов раздражения брюшины не было. Энтеральное питание усваивал, отмечались снижение аппетита, запоры в течение 1 нед. Мочеиспускание не контролировал, цвет мочи соломенно-желтый. По поводу стеноза чревного ствола больной консультирован сосудистым хирургом, от хирургической тактики решено воздержаться, рекомендовано консервативное лечение. При осмотре неврологом: сознание ясное, по шкале комы Глазго (ШКГ) — 15 баллов. Ориентация в месте, времени, собственной личности полная. Менингеальные симптомы отрицательные. Зрачки OD = OS, реакция на свет в норме. Движения глазных яблок в полном объеме. Лицо симметричное, язык по средней линии. Дизартрии, дисфонии, дисфагии не было. Мышечный тонус снижен. Тетрапарез по шкале оценки мышечной силы: в руках до 2 баллов в проксимальных и до 4 баллов в дистальных отделах, в ногах — до 3 баллов в проксимальных и дистальных отделах. Сухожильные рефлексы не вызывались. Нарушений чувствительности не было. Имелись нарушения тазовых функций по типу недержания мочи, запоров. Неврологом рекомендовано дообследование (электронейромиография (ЭНМГ), магнитно-резонансная томография (МРТ) головного мозга, шейного и грудного отделов позвоночника, исследование ликвора) для дифференциальной диагностики и уточнения генеза полинейропатии. При лабораторном обследовании обнаружены лейкоцитоз, анемия легкой степени и тромбоцитопения (лейкоциты 13,3·109/л, гемоглобин 12,3 г/дл, тромбоциты 114·109/л соответственно), повышение уровня панкреатической амилазы (77 Ед/л), гипоальбуминемия (30 г/л), электролитные нарушения (натрий 124 ммоль/л, калий 3,5 ммоль/л), повышение уровней воспалительных маркеров (С-реактивный белок 47,6 мг/л, прокальцитонин 7,4 нг/мл), удлинение активированного частичного тромбопластинового времени (40 с), снижение сывороточной концентрации железа (2,2 мкмоль/л) и фолиевой кислоты (1,9 нг/мл). При компьютерной томографии (КТ) органов грудной полости выявлены единичные очаги в нижних долях легких.
Начаты коррекция водно-электролитных нарушений (инфузии растворов натрия хлорида 0,9% и калия хлорида 4%) и анемии (фолиевая кислота, сульфат железа), эмпирическая антимикробная терапия (левофлоксацин), смешанное питание, профилактика стресс-язв (омепразол), профилактика тромботических осложнений (эноксапарин натрия), мероприятия по реабилитации в ОРИТ, включая механотерапию в прикроватных тренажерах, лечебную физкультуру (ЛФК), высаживание в прикроватное кресло.
В рамках дообследования при ЭНМГ обнаружена аксонально-демиелинизирующая (первично аксональная) моторно-сенсорная полинейропатия. При МРТ данных за стенозирование шейного и грудного отделов позвоночного канала, наличие объемных образований, очаговое поражение головного мозга не получено. Анализ ликвора от 30.01.18: бесцветный, прозрачный, цитоз 2 кл/мкл, глюкоза 3,9 ммоль/л, белок 0,2 г/л; вирусологические данные (вирус Эпштейна—Барр, цитомегаловирус, вирус простого герпеса 1-го и 2-го типов) — отрицательные. При бактериологическом исследовании крови, дистального конца удаленного венозного катетера и мокроты обнаружен рост золотистого стафилококка, в связи с чем с 30.01.18 левофлоксацин заменен на линезолид.
Учитывая результаты дообследования, состояние пациента расценили как проявление синдрома Гийена—Барре и кахексии с алиментарной недостаточностью вследствие перенесенного панкреатита, осложнившихся развитием нозокомиальной пневмонии, а также катетерассоциированной инфекции кровотока и тромбоза яремной и подключичной вен. 31.01.18 начат 5-дневный курс специфической терапии синдрома Гийена—Барре иммуноглобулином G — препаратом Октагам («Octapharma Pharmaceutica Production GmbH», Австрия) в дозе 0,4 мг на 1 кг массы тела в сутки.
Несмотря на проведенное лечение и ежедневные реабилитационные мероприятия, неврологическая симптоматика сохранялась на прежнем уровне. 05.02.18 усилились боли в конечностях. Учитывая длительный анамнез иммобилизации в ОРИТ, симптоматику расценили как проявления ПИТС, и к терапии добавлены флувоксамин и карбамазепин. Кроме того, сохранялись электролитные нарушения, и с целью исключения надпочечниковой недостаточности пациент дополнительно обследован: суточная экскреция натрия с мочой составила 87 ммоль/л, сывороточный уровень кортизола 19,7 мкг/дл. Учитывая полученные данные, к терапии добавили гидрокортизон. В связи с неэффективностью монотерапии линезолидом (повторный рост золотистого стафилококка в крови 05.02.18, сохраняющаяся высокая температура тела), антимикробная терапия усилена цефазолином. При повторном дуплексном сканировании отмечены сохраняющиеся тромбозы подключичной и внутренней яремной вен, что, вероятно, и служило причиной стойкой бактериемии, несмотря на проводимую антимикробную терапию.
Для исключения инфекционного эндокардита выполнена чреспищеводная эхокардиография, при которой данных за наличие вегетаций на клапанном аппарате и в полостях сердца не получено. Во время эзофагогастродуоденоскопии обнаружен грибковый эзофагит, позже подтвержденный результатами микроскопии.
После начала терапии карбамазепином и флуконазолом 08.02.18 состояние пациента с отрицательной динамикой в виде снижения уровня сознания до оглушения (14 баллов по ШКГ), появления спонтанного нистагма, тошноты. При повторной МРТ головного мозга с целью исключения понтинного миелинолиза изменений не выявлено. На следующие сутки восстановилось ясное сознание, однако пациент стал эмоционально лабилен, начал жаловаться на нарастающие ноющие боли в конечностях и сердце, при этом изменений на электрокардиограмме не было.
По совокупности всех клинико-лабораторных и инструментальных данных у пациента заподозрена острая порфирия, что потребовало специального лабораторного подтверждения. 09.02.18 в анализе мочи обнаружен порфобилиноген (ПБГ) 200,6 мг/л, δ-аминолевулиновая кислота (АЛК) 145,6 мг/л. Пациент переведен в отделение реанимации Национального медицинского исследовательского центра гематологии, где находился в течение 25 сут. В рамках патогенетической терапии острой порфирии проводилась постоянная инфузия 20% раствора глюкозы в объеме 1000 мл/сут, проведен курс аргинатом гема (препарат Нормосанг, «Orphan Europe S.a.r.L.», Франция) 125 мг/сут. В динамике в результате проводимой терапии отмечено снижение концентрации ПБГ с 200 до 31 мг/л, АЛК с 146 до 23 мг/л. Пациенту продолжена антибактериальная терапия стафилококковой инфекции (очаговая двусторонняя пневмония, инфекция кровотока). Диагностирован пиелонефрит, вызванный Pseudomonas aeruginosa.
Проводилась терапия тромбозов вен нижних конечностей и яремной вены справа. В связи с сохраняющейся алиментарной недостаточностью пациент получал смешанное питание. Течение заболевания в отделении реанимации осложнилось развитием симптоматического психоза, в связи с чем проводилась терапия галоперидолом с положительным эффектом. По данным лабораторных исследований выявлены признаки фолиеводефицитной и железодефицитной анемии, проводилась заместительная терапия фолиевой кислотой. На фоне коррекции гипонатриемии удалось стабилизировать уровень натрия в пределах 132 ммоль/л (минимальный уровень достигал 120 ммоль/л). Ежедневно проводились занятия кинезиотерапией и ЛФК. 04.04.18 пациент был выписан. Его состояние расценивалось как средней степени тяжести и обусловлено течением основного заболевания, сохраняющимся вялым парезом конечностей, алиментарной недостаточностью, анемией сочетанного генеза, тромботическими осложнениями.
Анализы и диагностика
В целях диагностики используются специальные методы лабораторного определения порфиринов/их предшественников в кале и моче, и в крови — для оценки активности ферментов.
Так, для болезни Гюнтера в клинико-биохимических анализах крови характерны признаки гемолиза — повышение сывороточного железа/непрямого билирубина, пойкилоцитоз, ретикулоцитоз, сфероцитоз, анизоцитоз, увеличение печеночных трансаминаз. Для пациентов с острыми порфириями характерно снижение уровня натрия/глюкозы, при поздней кожной порфирии— увеличение ферритина/сывороточного железа, положительные маркеры на вирус гепатита С.
При постановке диагноза важен семейный анамнез: наличие у близких родственников заболевания, а также обстоятельства начала заболевания (беременность, менструации, инсоляция, прием алкоголя/лекарств, голодание, инфекции и др.). Для подтверждения диагноза определяются уровни различных ферментов цикла биосинтеза гема в плазме, эритроцитах, лимфоцитах.
Из инструментальных методов могут назначаться КТ/УЗИ органов брюшной полости, КТ/рентгенография грудной клетки, ЭКГ, ЭЭГ головного мозга и др.
Диагностические мероприятия
Диагностика порфирии выполняется гематологом. Врач осматривает ребенка или взрослого и включает в анамнез все симптомы, которые могут указывать на нарушения в процессе синтеза гема. Пациенту предстоит ответить на вопросы о принимаемых медикаментах, режиме питания, перенесенных инфекционных заболеваниях. Девушек врач расспрашивает о стабильности менструального цикла, беременностях и абортах.
Следующим этапом диагностики становятся лабораторные тесты. Пациентам назначаются:
- общеклинический и биохимический анализы крови;
- проба Эрлиха;
- исследование концентрации ферментов в крови;
- ПЦР-тесты на гепатит;
- молекулярно-генетические исследования.
При наличии соответствующих показаний ребенок или взрослый посещает консультацию дерматолога, нефролога и гастроэнтеролога. Дифференциальная диагностика позволяет врачам исключить из анамнеза пациента неврологические и психиатрические патологии.
Профилактика
Специфическая профилактика отсутствует. С целью минимизации приступов рекомендуется:
- Избегать/минимизировать воздействие порфириногенных факторов (режимный образ жизни, избегать стрессов, приема потенциально опасных лекарств, употребления алкогольсодержащих напитков, голодания/строгих диет, пребывания на солнце).
- Пациенты старше 50 лет должны проходить дважды в год обследование для оценки состояния печени.
- Рациональное трудоустройство.
- Проведение профилактического лечения/диспансеризация больных.
- При наличии в семье больного порфирией необходимо проводить ДНК-диагностику (выявление генетических мутаций) и определять активность различных ферментов цикла синтеза гема.
Лечение порфирии
Лечением порфирий занимается гематолог. Так заболевание носит генетический характер, терапия сводится лишь к снижению симптомов.
Причины и симптомы порфирии
При эритропоэтических порфириях на первый план выходит защита от солнца: максимально закрытая одежда, шляпы с широкими полями, солнцезащитные кремы. Для повышения фотозащиты могут быть рекомендованы препараты бета-каротина, никотиновой кислоты.
Чтобы отсрочить развитие цирроза при печеночных порфириях, пациентам назначают гепатопротекторы — например, препараты урсодезоксихолевой кислоты (Урсосан), которые повышают устойчивость гепатоцитов (клеток печени) к внешним токсическим воздействиям и таким образом снижают активность апоптоза (гибели клеток).
Для уменьшения уровня порфиринов в организме могут быть рекомендованы кровопускания и прием противомалярийных препаратов. В качестве поддерживающей терапии назначают витамины, АТФ и кокарбоксилазу.
Последствия и осложнения
Поскольку порфирии за исключением поздней кожной порфирии являются наследственно обусловленными их излечение полностью невозможно. При наличии у пациента 2-3 и более приступов в год может длительно сохраниться неврологический дефицит в виде вялых периферических парезов. В случаях тяжелого течения возможны осложнения в виде бульбарного синдрома, гипонатриемии, паралича дыхательной мускулатуры с высокой угрозой летального исхода. При порфириях с поражением кожных покровов — риск развития склеродермоидных процессов.
Общая информация
Порфириновая болезнь диагностируется сравнительно редко: российские врачи выявляют не более 12 случаев на 100 тысяч человек. Различные формы патологии получают распространение в отдельных регионах Земли. Так, признаки поздней кожной порфирии часто выявляется у жителей Южной Африки (1 случай на 800 человек). Острый перемежающийся тип заболевания характерен для жителей Северной Европы (1 случай на 1000 человек). Мужчины и женщины одинаково часто страдают от различных форм синтеза гема.
Список источников
- Кузнецова Н.П., Панков Б.С., Чубарова А.С. и др. Порфирии. М., 1981. с. 66–14.
- Пустовойт Я.С., Пивник А.В., Карпова И.В. Клиника, диагностика и лечение порфирий // Пособие для врачей. Москва, 2003 г.
- Воробьёв А, Кравченко С, Кременецкая А, Карпова И, Пустовойт Я. Острая перемежающаяся порфирия: проблемы диагностики и лечения // Врач. 2003 г. №2, стр. 8-13.
- Диагностика и лечение острых порфирий: Клинические рекомендации национального гематологического сообществ/ под ред. Пустовойт Я.С., Кравченко С.К., Шмакова Р.Г., Савченко В.Г. — 2022.
- Руководство по гематологии в 3 томах/ под ред. Воробьева А.И. – 2005 – Т.3.