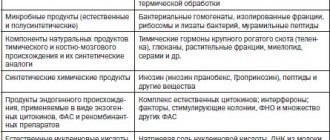
Porphyrias are a group of genetically determined diseases associated with the heme biosynthesis cycle (Fig. 1). The development of any nosological form of porphyria is a consequence of a decrease in the activity of one of the enzymes of the heme biosynthesis cycle, as a result of a mutation in a pathognomonic gene. According to the modern classification, porphyrias are divided into:
- according to the place of predominant disturbance of porphyrin metabolism - for erythropoietic and hepatic porphyrias (Table 1);
- according to clinical manifestations - into acute forms that occur with predominantly damage to the nervous system, and forms that occur with skin damage (see Table 1).
Table 1
Classification of porphyrias
| Forms of porphyria | Porphyria |
| Erythropoietic forms | Congenital erythropoietic porphyria |
| Erythropoietic protoporphyria | |
| Hepatic forms | X-linked dominant protoporphyria |
| Porphyria due to d-ALA dehydratase deficiency | |
| Acute intermittent porphyria | |
| Congenital coproporphyria | |
| Variegate porphyria | |
| Porphyria cutanea tarda | |
| Acute forms of porphyria | X-linked dominant protoporphyria |
| Porphyria due to d-ALA dehydratase deficiency | |
| Acute intermittent porphyria | |
| Congenital coproporphyria | |
| Variegate porphyria | |
| Forms that occur with damage to the skin | X-linked dominant protoporphyria |
| Porphyria cutanea tarda | |
| Congenital coproporphyria | |
| Variegate porphyria | |
| Congenital erythropoietic porphyria | |
| Erythropoietic protoporphyria |
Differences in clinical manifestations in various nosological forms depend on the level of the heme biosynthesis cycle, at which the enzyme functions with reduced activity, which determines which fraction of porphyrins will predominate in the resulting excess of metabolites. If heme metabolism is blocked at a high stage of the cycle, accumulation of isomers of porphyrins proper occurs, which are tropic to the dermis and lead to photodermatosis. With an enzymatic defect located at the initial stages of the heme biosynthesis cycle, porphyrin precursors (porphobilinogen and d-aminolevulinic acid, d-ALA), which have a neurotoxic, demyelinating effect, leading to sensory-motor polyneuropathy, will predominate among the metabolites.
Rice. 1. Scheme of the heme biosynthesis cycle indicating possible points of realization of the porphyrinogenic effects of drugs.
These effects are mediated through possible pathways (1a-1c; 2d-2e) influencing the synthetic activity of the cycle.
Regardless of the tissue affiliation or the characteristics of the clinical course, all forms of porphyria are characterized by common links in pathogenesis. For clinical manifestation of any form of porphyria it is necessary that there is an excessive accumulation of porphyrins, usually resulting from (see Fig. 1):
1. Increased activity of δ-ALA synthetase, the first enzyme in the heme biosynthesis cycle, the reasons for which may be:
a) the presence of a mutation in the δ-ALA synthetase gene, leading to an increase in its activity;
b) a direct inducing effect on the enzyme of factors of an endogenous or exogenous nature, for example, an increase in the level of endogenous progesterone;
c) increased consumption of heme, the final product of the heme biosynthesis cycle, leading to activation of δ-ALA synthetase according to the feedback principle.
2. Partial or complete decrease in the activity of one of the enzymes of the heme biosynthesis cycle, leading to the accumulation of porphyrin metabolism products. The following reasons for decreased enzyme activity are possible:
d) the presence of a pathological mutation in the enzyme gene;
e) an inhibitory effect on the enzyme of exogenous or endogenous factors (for example, an isolated decrease in the activity of uroporphyrinogen decarboxylase, leading to the debut of a sporadic form of porphyria cutanea tarda).
The action of known porphyrinogenic factors (Table 2), leading to an escalation of porphyrin synthesis, can be mediated through the pathways described above. One of the most significant porphyrinogenic factors are drugs. Taking porphyrinogenic drugs is the cause of the development of attacks of acute porphyria (AP) in 18% of cases (own unpublished data). Drug-induced attacks of AP are more severe than attacks caused by other factors. According to our data, when AP was provoked by drugs, a severe course of the disease was observed in 100% of cases, and 45% of patients required long-term, more than 2 months, hospital treatment. In patients with nosological forms that occur with damage to the skin, taking porphyrinogenic drugs leads to an escalation of porphyrin synthesis and the subsequent manifestation of photodermatosis.
table 2
Porphyrinogenic factors
| Porphyrinogenic factors |
| Starvation |
| Infections |
| Alcohol |
| Taking medications |
| Reproductive function in women |
| Insolation |
| Mental stress |
| Physical overload |
The porphyrinogenicity of drugs is realized through mechanisms that influence different parts of porphyrin metabolism (see Fig. 1).
The most common trigger mechanism is the activation of the cytochrome P450 system. Half of the drugs undergo oxidative metabolism in the liver through a detoxification system involving cytochrome P450 (CYP) enzymes. They catalyze reactions of hydroxylation, epoxidation, N-, S-, O-dealkylation, N-oxidation, sulfoxidation, dehalogenation and other reactions leading to the formation of water-soluble metabolites. There are 30–100 enzyme subtypes in the CYP system. Isoenzymes involved in the metabolism of xenobiotics belong to CYP families 1, 2, 3 and 4. The production of CYP apoproteins at the level of gene transcription is coordinated with the production of the ALAS1 enzyme, which is necessary for heme, which is part of the CYP holoenzyme. Therefore, drugs that induce CYP activate transcription of the ALAS1 gene. In humans, drug-induced transcription of almost all CYP enzymes is mediated by the constitutively active receptor (CAR) and the pregnane xenobiotic receptor (PXR). A drug that is a ligand for PXR or CAR should immediately be considered a potential porphyrinogenic drug. For the porphyrinogenic effect of a drug to manifest, the volume of CYP apoenzyme induced by it must be sufficient to cause ALASl catalysis in mitochondria to produce the heme necessary for the formation of the holoenzyme, and also sufficient to cause a significant consumption of heme in hepatocytes. In addition to the amount of substance initiating this process, it is of great importance which subclasses of CYP will be induced. Almost all porphyrinogenic drugs are inducers of CYP3A4 and/or CYP2C9, for example antiepileptic drugs, the calcium channel blocker nifedipine, sulfadiazine, sulfamethoxazole and rifampicin, the fungicide ketoconazole, progesterone, testosterone. At the same time, powerful inducers of CYP2E1, 2C19 and 1A1/1A2, for example, cimetidine, acetylsalicylic acid and omeprazole, do not pose a danger to patients with AP. Some porphyrinogenic substances are inducers of many microsomal liver enzymes, including CYP isoenzymes and uridine diphosphate glucuronosyltransferases (phenobarbital, phenytoin, carbamazepine). They affect more than one system, which is why they are called multifunctional inductors. Their porphyrinogenicity is explained by intense transcription of the ALAS1 gene, triggered by the simultaneous induction of several CYP subclasses.
Drugs can have a direct effect on the synthetic activity of the first enzyme of the heme biosynthesis cycle, in particular hormonal drugs. For example, drugs such as the desogestrel derivative - regulon.
Drugs can inhibit the activity of enzymes in the biosynthetic chain. For example, the already mentioned oral contraceptives.
In addition to the listed mechanisms of action of drugs, there are also different ways to regulate the activity of the heme biosynthesis system, both as a whole and its individual parts, which is reflected in daily fluctuations in the excretion of porphyrins and precursors in the urine in patients with AP. This, in turn, may temporarily create more favorable conditions for the implementation of drug-induced porphyrinogenic effects.
For porphyrinogenicity of drugs to manifest, it is necessary that their concentration in hepatocytes is not lower than micromolar. Therefore, at daily doses in the milligram range and below, the effect on the liver will be insufficient to trigger the porphyrinogenic transcription of ALAS1. Therefore, not only the fact of prescribing a porphyrinogenic drug is important, but also its dose, as well as the route of administration. When taken orally, for example, aminoglycoside antibiotics, the systemic exposure is insignificant and does not cause an attack of AP. In addition, after oral administration, the drug is metabolized in the intestinal wall, which also reduces the amount of unmetabolized substance entering the liver through the portal vein. There are significant interindividual differences in the extent of xenobiotic metabolism in the intestinal wall, making it difficult to predict liver burden based on dose information. Examples of such drugs are salbutamol, verapamil and lidocaine. The drugs enter the bloodstream when applied to the surface of the skin, local use: applied to the surface of the skin, conjunctival sac, cornea or mucous membrane of the vagina, rectum, mouth or nose. The extent of drug absorption through the rectal mucosa is usually unpredictable. The systemic effect of drugs that are not administered directly into the vascular bed depends to a greater extent on the vascularization of the tissue, i.e. from the injection site. Subcutaneous and single perineural injections are equivalent in this regard. Inhalation administration of drugs, such as liquid vaporized anesthetics and respiratory medications, can also introduce lipophilic substances into the blood. Water-insoluble substances used in the form of an aerosol powder, such as sodium cromoglycate, are poorly absorbed and their systemic exposure can be neglected. The rate of systemic resorption from the cerebrospinal fluid depends on the degree of binding of the drug to proteins, which varies for different substances. With epidural administration, the concentration of local anesthetic in the cerebrospinal fluid is much higher than in the plasma, and with intrathecal administration, the concentration in plasma may not reach the detection limit. Thus, absorption and distribution effects may reduce the porphyrinogenicity of drugs.
Issues of selecting drug therapy for AP are relevant not only during the treatment of an acute attack of the disease, the main goal of which is to prevent repeated attacks during treatment, but also at the outpatient stage. According to our data, up to 40% of cases of outpatient visits by patients with AP are dictated by the emergence of issues related to drug therapy, such as:
— selection of drugs for the treatment of competing diseases and delayed complications of AP;
— selection of medications during pregnancy;
— selection of safe drugs for anesthesia during planned surgical interventions.
Thus, information about the porphyrinogenicity of various drugs is important. And here two extremes are possible. On the one hand, ignorance, underestimation or ignoring the porphyrinogenicity of a drug when prescribing it can lead to the development of an acute attack of porphyria. On the other hand, refusing to prescribe a drug that is known to be porphyrinogenic for health reasons can cause more harm to the patient than its use (for example, prescribing oncological drugs, in some cases antibiotic drugs, etc.). In these cases, porphyrinogenic drugs can be prescribed simultaneously with heme arginate, which will reduce the production of toxic metabolites of porphyrin metabolism, or an alternative route of administration can be chosen, which will reduce their systemic effect (inhalation, instillation, etc.).
Unfortunately, at present, attempts to predict the porphyrinogenicity of a particular drug only on the basis of knowledge about its metabolism are not always successful. Therefore, lists of porphyrinogenic and safe drugs for porphyria are being created based not only on the participation of the cytochrome P450 system in their metabolism, but also on individual clinical reports and observations. These lists are published in print (https://www.porphyriafoundation.com/drug-database, https://www.porphyria-europe.com/02-for-healthcare/Abstracts/AbstractList.asp, https://porphyriadrugs. com) in the form of databases located on the Internet.
The Federal State Budgetary Institution Hematological Research Center of the Ministry of Health of the Russian Federation (FSBI National Medical Research Center for Hematology of the Ministry of Health of the Russian Federation) has accumulated one of the largest clinical experiences in the treatment and monitoring of patients with AP. Over a nearly 20-year period, the Federal State Budgetary Institution National Medical Research Center for Hematology of the Ministry of Health of the Russian Federation observed 163 patients with this orphan disease, many cases of provocation of AP by various drugs were noted, and our list of porphyrinogenic drugs does not always coincide with the lists proposed by foreign authors. In some cases, up to 15% of drugs that the authors (https://porphyriadrugs.com/search/?q=cefo, https://porphyriadrugs.com/search/?q=lido) consider safe, according to our data, provoke attacks of AP . For example, drugs such as ketonal, lidocaine, ceftriaxone, according to the website (https://porphyriadrugs.com) are considered safe for AP; according to our data, as well as according to a number of other authors, these drugs have a strong porphyrinogenic effect. Therefore, we have developed our own Internet version of the register of drugs that are dangerous and safe for patients with porphyria, accessible to all doctors, which is posted on the website of the Federal State Budgetary Institution National Medical Research Center for Hematology of the Ministry of Health of the Russian Federation. The database being created was based on an electronic reference module for medicines, kindly provided by the editors of the Vidal publication. More than four thousand names of medicines were analyzed, from which the drugs used in the treatment of patients at the Federal State Budgetary Institution National Medical Research Center for Hematology of the Ministry of Health of the Russian Federation were selected. All drugs, depending on their porphyrinogenicity, are highlighted in different colors. Drugs with high porphyrinogenicity are highlighted in red, safe drugs are highlighted in green. The names of drugs whose porphyrinogenicity is not fully understood are colored yellow. It is better to avoid taking them if a safe analogue is available. The database can be searched both by proprietary names of medicines and by international non-trade names (Fig. 2).
Rice. 2. Screenshot of the registry, based on the drug database module of the Vidal publication.
Approved drugs are highlighted in green, drugs with high porphyrinogenic activity are highlighted in red, and drugs with insufficient information about porphyrinogenicity are highlighted in yellow.
When prescribing a particular drug, it is necessary to take into account that, for vital indications, known porphyrinogenic drugs can be prescribed simultaneously with heme arginate. The degree of risk of death from the disease should not exceed the degree of risk of developing an attack of AP when using a porphyrinogenic drug. First of all, this concerns the treatment of shock conditions, the provision of resuscitation aid, and the treatment of oncological diseases.
Porphyrinogenic drugs can be used locally (applications, irrigation, inhalation). Since many infectious processes are local in nature, the successful use of porphyrinogenic antimicrobial agents is acceptable.
Vaccines, serums, and monoclonal antibodies are considered non-porphyrinogenic, but the immunological reactions of the body that occur during their administration can provoke an attack of AP.
The proposed register of porphyrinogenic drugs is advisory in nature. Only the attending physician can make a final decision on prescribing a particular drug after analyzing the course of the disease. As additional information becomes available, it is planned to update the register and make additions to it.
MEDICAL CENTER
- Level of circulating interferon (serum interferon).
- Spontaneous production of interferon in vitro.
- Induced synthesis of alpha-interferon in vitro.
- Induced synthesis of interferon gamma in vitro.
Additional tests:
- Determination of sensitivity to interferon drugs (No. 1044 - Ingaron, No. 1045 - Intron, No. 1047 - Reaferon, No. 1048 - Realdiron, No. 1049 - Roferon).
- Determination of sensitivity to interferon inducers (No. 1050 - Amiksin, No. 1051 - Kagocel, No. 1052 - Neovir, No. 1053 - Ridostin, No. 1054 - Cycloferon).
- Determination of sensitivity to immunomodulators (No. 1055 - Galavit, No. 1056 - Gepon, No. 1057 - Immunal, No. 1058 - Imunofan, No. 1059 - Immunomax, No. 1060 - Likopid, No. 1061 - Polyoxidonium, No. 1062 - Taktivin, No. 1063 - Thymogen, No. 1066 - Imunorix, No. 1148 - Panavir, No. 1064 - Isoprinosine).
Interferons (IFNs) are the most important component of the body’s innate nonspecific defense against infections (the name interferons comes from their ability to interfere with viral infection of cells).
This is a family of proteins of local (autocrine and paracrine) regulation that are capable of activating intracellular processes and intercellular interactions that provide resistance to viral infections, enhance innate and acquired immune responses, modulate the processes of development and death of normal and tumor cells. The body's resistance to viral infections and a number of other diseases largely depends on the activity of a group of genes of the interferon system. Interferon preparations are widely used in medicine. The effects of interferons are indirect - activation of specific receptors by interferons causes a cascade of cellular processes leading to the induction of specific interferon-stimulated genes encoding the synthesis of many proteins, which provide the antiviral effects, antitumor and antiproliferative effects of interferons. Proteins induced by interferons include: enzymes, transcription factors, cell surface glycoproteins, cytokines, chemokines and other factors, the effect of which continues to be studied. The production of interferons by cells is transient, temporary - normally “silent” interferon genes are induced under the influence of products of viral and microbial origin and chemical inducers.
Interferons are divided into three types (α, β and γ), which are associated with specific functions and specific producing cells. Interferons α and β, despite significant structural differences, have common receptors and similar functions. Together they are also called type I interferons, or acid-stable interferons, in contrast to interferon-γ, which has its own receptors and partially different functions (it is also known as interferon II, or acid-labile interferon).
Interferon α (more than 20 of its subtypes have been identified) is the main interferon that is synthesized in a virus-induced leukocyte culture. The main producers of IFN-α are plasmacytoid dendritic cells; monocytes make a significant contribution to the IFN-α-producing ability of the blood. Its main functions are antiviral activity and activation of natural killer cells.
Interferon β is the main interferon produced by double-stranded RNA-induced fibroblast culture. Its main producers are fibroblasts, epithelial cells and macrophages; its main function is antiviral activity.
Interferon γ is the main interferon produced by immunologically stimulated (mitogens or antigens) lymphocyte cultures. The main producing cells of IFN-γ are T-lymphocytes. The main function of interferon gamma is immunoregulation (including activation of macrophages, enhancement of the Th1 response, induction of the expression of major histocompatibility complex type II antigens on antigen-presenting cells, etc.); like other interferons, it exhibits antiviral and antiproliferative activity. All animal cells are capable of producing interferons; certain cells (leukocytes and fibroblasts) can produce more than one type - both IFN-α and IFN-β.
Studying the parameters of interferon status allows us to identify insufficiency of the interferon system. Assessment of detected changes can serve as a guide in the diagnosis, treatment and prognosis of diseases of both viral and non-viral etiology. Healthy people are characterized by low levels of serum interferon and high values of induced interferon synthesis. Stress and acute viral infections, allergic conditions are accompanied by an increase in the level of circulating interferon and a decrease in the level of induced production of alpha and gamma interferons by leukocytes. In bronchial asthma and urticaria, the level of circulating interferon correlates with the severity of the disease.
Chronic viral infections (herpes, hepatitis), multiple sclerosis are accompanied by suppression of all indicators of interferon status. Autoimmune diseases (systemic lupus erythematosus, rheumatoid arthritis) are characterized by suppression of inducible production of interferon alpha. Acute lymphocytic leukemia and malignant formations are accompanied by suppression of induced interferon gamma production. The results of the study of interferon status should be considered in conjunction with other laboratory and clinical anamnestic data. A decrease in the production of alpha and gamma interferon, which can be both a cause and a consequence of acute and chronic viral diseases, indicates a congenital or acquired deficiency of the interferon system and can be considered as an indication for interferon-stimulating therapy. Normalization of interferon status indicators usually coincides with the recovery process. In people over 50 years of age, insufficiency of the interferon system is relatively more common. The study of interferon status parameters with determination of sensitivity to drugs is used to select effective therapy when using exogenous interferon drugs, interferon inducers and immunomodulators.
Interferon preparations
- Ingaron is a recombinant human interferon-γ.
- Intron - recombinant human interferon-α-2b.
- Reaferon is a recombinant human interferon-α-2.
- Realdiron is a recombinant human interferon-α-2b.
- Roferon is a recombinant human interferon-α-2a.
Interferon inducers
- Amiksin (international non-patent name - tilorone): dihydrochloride 2,7-bis-[2(diethylamino)-ethoxy]-fluorene-9-OH-dihydrochloride.
- Kagocel: active substance - sodium salt of the copolymer (1-4)-6-0-carboxymethyl-bD-glucose, (1-4)-bD-glucose, (21-24)-2,3,14,15,21, 24,29,32-octahydroxy-23-(carboxy-methoxymethyl)-7,10-dimethyl-4,13-di(2-propyl)-19,22,26,30,31-pentaoxaheptacyclo [23.3.2.216.05.28 .08.27.09.1В.012.17] dotriaconta-1,3,5(28),6,8(27),9(18),10,12(17),13,1-decaene.
- Neovir – 2-(9-oxo-9,10-dihydroacridin-10-yl) sodium acetate.
- Ridostin is a mixture of sodium salts of double-stranded and single-stranded RNA.
- Cycloferon – meglumine acridone acetate.
Immunomodulators
- Galavit is a phthalhydrazide derivative.
- Hepon is a synthetic peptide consisting of 14 amino acid residues.
- Immunal is a preparation of Echinacea purpurea juice.
- Immunofan is a hexapeptide (arginyl-alpha-aspartyl-lysyl-valyl-tyrosyl-arginine).
- Immunomax is an acidic peptidoglycan with a molecular weight of 1,000 - 40,000 kDa.
- Lykopid - active substance - glucosaminylmuramyl dipeptide - 4-O-(2-acetylamino-2-deoxy-beta-D-glucopyranosyl)-N-acetylmuramyl]-L-alanyl-D-alpha-glutalamide.
- Polyoxidonium - international non-proprietary name / composition: Azoximer (Azoximer) - N-oxidized derivative of polyethylene piperazine.
- Taktivin is a complex of polypeptides from the thymus gland of cattle.
- Thymogen is a polypeptide from the thymus gland of cattle.
- Panavir is a purified extract of shoots of the Solanum tuberosum plant, the main active ingredient is a hexose glycoside consisting of glucose, rhamnose, arabinose, mannose, xylose, galactose, uronic acids.
ANTIVIRAL DRUGS WITH AN EXTENDED SPECTRUM OF ACTIVITY
MODERN ANTIMICROBIAL CHEMOTHERAPY
MODERN ANTIMICROBIAL CHEMOTHERAPY
L.S. Strachunsky, S.N. Kozlov. Guide for doctors
| Content | ANTIBIOTIC.ru |
Antiviral drugs
RIBAVIRIN
Virazol, Rebetol
It has a wide spectrum of activity against many DNA and RNA viruses and is highly toxic. The mechanism of antiviral action is not fully understood.
Activity spectrum
Of clinical importance is activity against respiratory syncytial virus, as well as viruses that cause Lassa fever, hemorrhagic fever with renal syndrome and hepatitis C (in combination with interferon-alpha).
Pharmacokinetics
Bioavailability when taken orally is 35-45%. When administered by inhalation, high concentrations are observed in respiratory secretions and significantly lower concentrations in plasma. With repeated administrations it can accumulate in red blood cells. Penetrates through the BBB. Metabolized in the liver, excreted in the urine. T1/2 - 30-60 hours, increases with renal failure.
Adverse reactions
- Local - rash, irritation of the skin, mucous membranes of the eyes and respiratory tract, bronchospasm (noted by both patients and medical staff
when using an aerosol dosage form). Ribavirin may crystallize in the respiratory tract and endotracheal tubes. - Hematotoxicity - anemia, lymphocytopenia (in AIDS patients); hemolytic anemia (usually by 4 weeks), reversible, does not require specific treatment, normalization of hemoglobin occurs with a temporary dose reduction.
- Neurotoxicity - headaches, fatigue, irritability, insomnia.
- Gastrointestinal tract - metallic taste in the mouth, abdominal pain, flatulence, nausea.
- Teratogenic effect.
Indications
- Infections caused by RSV (serologically confirmed): severe bronchiolitis and pneumonia in newborns and young children at risk (congenital heart defects, immunodeficiency, bronchopulmonary dysplasia), as well as those associated with severe cystic fibrosis or pulmonary hypertension.
- Lassa fever.
- Hemorrhagic fever with renal syndrome.
- Hepatitis C (in combination with interferon-alpha or peginterferon alfa).
Contraindications
Absolute
- Pregnancy.
- End-stage renal failure.
- Anemia.
- Hemoglobinopathies.
- Severe heart failure.
Relative
- Uncontrolled hypertension.
- Elderly age.
Warning
Due to its teratogenic effects, it is contraindicated during pregnancy and poses a danger to medical staff if pregnant.
All women receiving ribavirin (and if their partners are receiving it) should be protected against pregnancy during the entire course of therapy and for 4 months after the end of treatment. The pregnancy test must be repeated monthly, as well as for 4 months after the end of treatment. If pregnancy occurs during this period, the patient must be warned about the high risk of teratogenic effects.
In order to “protect” medical staff, inhaled administration of ribavirin is allowed only with the use of a special nebulizer.
Before using ribavirin, mandatory serological confirmation of the presence of RSV infection is required, as well as determination of HCV RNA by polymerase chain reaction (in patients with hepatitis C).
Dosage
Adults
For Lassa fever and hemorrhagic fever: intravenously - the first dose is 2.0 g, then 1.0 g every 6 hours for 4 days and then 0.5 g every 8 hours for 6 days.
For hepatitis C: orally 1.0-1.2 g per day for 12 months.
Newborns and children
For RSV infection: inhalation (using a nebulizer) 20 mg/ml (6.0 g in 300 ml of sterile water) for 18 hours a day, course of treatment - 3-7 days.
Release form
Bottles of 6.0 g of powder for preparing a solution for infusion; capsules 0.2 g.
LAMIVUDINE
Zeffix, Epivir TriTC
Active against retroviruses and hepatitis B virus.
Pharmacodynamics
In cells affected by the virus, it is activated, turning into lamivudine triphosphate, which inhibits hepatitis B virus DNA polymerase and HIV reverse transcriptase. There have been cases of development of hepatitis B virus resistance.
Pharmacokinetics
Bioavailability when taken orally is 86-88%. Distributed into many tissues and secretions, passes through the BBB. Excreted by the kidneys. T1/2 - 5-7 hours, with renal failure it may be extended.
Adverse reactions
Generally well tolerated. In rare cases, it causes lactic acidosis and hepatomegaly with steatosis, which may be associated with impaired mitochondrial function.
Indications
- Chronic hepatitis B.
Warning
With monotherapy, resistance to lamivudine of both the hepatitis B virus and HIV can develop quite quickly if double infection occurs.
Dosage
Adults
Orally - 0.1 g once a day for a year; in HIV-infected patients - 0.15 g every 12 hours.
Release form
Tablets 0.1 g.
Interferons are a group of biologically active proteins synthesized by the cell during the protective reaction. Interferon is secreted into the extracellular fluid and acts on other cells through receptors, increasing resistance to intracellular microorganisms, primarily viruses. Interferons do not have specificity and inhibit the replication of various viruses. The main mechanism of the antiviral action of interferon is to suppress the synthesis of viral proteins.
According to their structure and biological properties, interferons are divided into three types: α, β, γ
. According to the method of production, leukocyte, lymphoblastoid and recombinant interferons are distinguished. Recombinant alpha interferons are most widely used as antiviral drugs. In recent years, pegylated interferons (peginterferons) have been developed, obtained by adding polyethylene glycol and having a higher half-life and clinical efficacy.
INTERFERON-ALPHA: GENERAL PROPERTIES
Pharmacokinetics
Being a protein, interferon-alpha is destroyed in the gastrointestinal tract, therefore it is used only parenterally. When administered intramuscularly, the bioavailability is 80%, the maximum concentration in the blood is achieved on average after 3.8 hours. Low concentrations have been noted in the secretions of the respiratory tract, eye tissues, and the central nervous system. It is rapidly inactivated in the kidneys, and to a lesser extent in the liver. T1/2 - 2-4 hours, does not change with renal failure. Peginterferon alfa has a longer T1/2.
Adverse reactions
Adverse reactions of recombinant interferon-alpha are dose-dependent.
Early
(usually in the first week of treatment).
- The flu-like syndrome, manifested by fever, myalgia, and soreness of the eyeballs, usually disappears after 4-5 injections and does not require dose reduction or discontinuation of the drug.
Preventive measures:
prescribing paracetamol before administering interferon.
Late
(at 2-6 weeks of therapy, often causes interferon withdrawal).
- Hematotoxicity - anemia, thrombocytopenia, agranulocytosis.
- Neurotoxicity - drowsiness, lethargy, depression, less often convulsions.
- Cardiotoxicity - arrhythmias, transient cardiomyopathy, arterial hypotension.
- Autoimmune thyroiditis.
- Hyperlipidemia.
- Alopecia, skin rashes. Control measures:
control of hematopoiesis, levels of liver enzymes, electrolytes, ECG.
Drug interactions
Interferon alpha inhibits microsomal liver enzymes (cytochrome P-450), and therefore can disrupt the metabolism of many drugs (theophylline, etc.), increasing their concentration in the blood.
Due to the risk of adverse reactions from the central nervous system, narcotic, hypnotic and sedative drugs should be used simultaneously with interferon alpha.
Indications
- Chronic hepatitis B (in the presence of viral replication: HBV, DNA, HBeAg, in blood serum) and elevated levels of transaminases.
- Acute hepatitis C.
- Chronic hepatitis C (HCV RNA in blood serum), elevated levels of transaminases.
Contraindications
Absolute
- Psychosis (at the time of treatment or in history).
- Severe depression.
- Neutropenia or thrombocytopenia.
- Severe heart pathology.
- Decompensated cirrhosis of the liver.
- Uncontrollable seizures.
- Organ transplantation (except liver).
Relative
- Autoimmune diseases.
- Uncontrolled diabetes.
Dosage
Adults
Chronic hepatitis B
5 million IU daily or 10 million IU 3 times a week for 4-6 months.
Acute hepatitis C
High dose regimen - 10 million IU daily until transaminases normalize, then 3 million IU 3 times a week for 6 months.
Medium dose regimen: 5 million IU 3 times a week for 2 months, then 3 million IU 3 times a week for 4-10 months.
If tolerance is poor, switch to a low dose regimen of 3 million IU 3 times a week for 3-6 months.
Chronic hepatitis C
Monotherapy - 3 million IU 3 times a week for 12 months. In the absence of a response (preservation of HCV RNA in the blood serum), combination therapy is advisable after 3 months from the start of therapy.
Combination therapy:
- 1) Interferon alpha + ribavirin. For body weight ≤75 kg, interferon alpha 3 million IU 3 times a week, ribavirin 1 g/day (2 capsules in the morning + 3 capsules in the evening). For body weight >75 kg, interferon alpha 3 million IU 3 times a week, ribavirin 1.2 g/day (3 capsules in the morning + 3 capsules in the evening).
2) Peginterferon alfa-2b + ribavirin. For body weight <65 kg, peginterferon alfa-2b 1.5 gc/kg once a week, ribavirin 0.8 g/day. For a body weight of 65-85 kg, peginterferon alfa-2b 1.5 g/kg once a week, ribavirin 1 g/day. For body weight >85 kg, peginterferon alfa-2b 1.5 mg/kg once a week, ribavirn 1.2 g/day.
For chronic hepatitis C - 3 million IU 3 times a week for 3 months, with normalization of transaminase levels and a decrease in the concentration of HCV RNA - administration at the same dose for 9-15 months. If, after 3 months from the start of therapy, the ALT level remains elevated and HCV RNA continues to be detected, the dose can be increased to 5 million IU 3 times a week.
Children over 1 year old
The effectiveness and safety of interferons in children have not been fully established. Controlled studies completed to date have revealed the effectiveness of the following treatment regimens: chronic hepatitis B - 6 million IU/m2 body surface 3 times a week for 6 months; chronic hepatitis C - 3-5 million IU/m2 3 times a week for 12 months.
INTERFERON ALPHA PREPARATIONS
Recombinant interferons
All commercial drugs in this group are a recombinant form of human α2-interferon, so their pharmacological action is similar. Depending on the amino acid content, interferon alpha-2a and interferon alpha-2b are distinguished, which do not differ significantly in clinical effectiveness and safety.
INTERFERON ALPHA-2a
Roferon-A, Reaferon
Release forms
Vials and ampoules of 3, 9 and 18 million IU ( Roferon-A
) and ampoules of 1 million IU (
Reaferon
) powder for the preparation of a solution for injection.
INTERFERON ALPHA-2b
Intron-A, Realdiron
Release forms
Bottles of 1, 3, 5 and 10 million IU ( Intron-A
) and ampoules of 1, 3 and 6 million IU (
Realdiron
) powder for the preparation of solution for injection.
PEGINTERFERON ALPHA-2b
PegIntron
A compound of interferon alfa-2b with polyethylene glycol (PEGylated interferon alfa-2b). It has a prolonged effect and higher therapeutic activity. Prescribed once a week. Recommended for the treatment of hepatitis C in persons who have contraindications to ribavirin, as well as in case of discontinuation of ribavirin due to the development of anemia.
Release forms
Vials of 50, 80 and 100 mcg of powder for the preparation of solution for injection.
| Copyright © 2000-2007 ANTIBIOTIC.ru Posted: 05/15/2004 |
The address of this page: https://www.antibiotic.ru/books/mach/mac0403.shtml
Last modified date: 05/24/2004 18:56
Possibilities of modern drugs in the prevention and treatment of ARVI and influenza
For citation. Shishkova V.N. Possibilities of modern drugs in the prevention and treatment of acute respiratory viral infections and influenza // RMZh. 2016. No. 6. pp. 395–400.
Acute respiratory viral infections (ARVI) are a pressing problem of modern medicine, especially during “transitional” climatic periods: winter-spring and autumn-winter, when the incidence of influenza and ARVI increases.
According to WHO, up to 100 million people worldwide fall ill with influenza and ARVI every year, of which almost 4 million die from complications [1]. More than 200 types of ARVI pathogens are known [2]. Traditionally and seasonally, there has been an increase in interest among the population and clinicians in the use of various anti-influenza and anti-cold drugs. Since the structure of seasonal morbidity in adults and children is dominated by acute diseases of viral etiology, occurring with intoxication syndrome, headache and joint pain, as well as damage to the upper respiratory tract, trachea, bronchi and having a certain similarity in pathogenesis and clinical symptoms, the drugs used in Most of them are antiviral, immunotropic and symptomatic agents. The abundance of names and forms of these groups of drugs, most of them over-the-counter, makes it almost impossible for the patient to make an independent correct choice. Let's try to deal with each group of drugs sequentially. So, in most cases, the root cause of ARVI is a respiratory viral infection, which has a whole range of adverse effects, but the main reasons for its development, as a rule, are impaired mucociliary clearance and a decrease in local mucosal immunity. A direct consequence of this circumstance is the proliferation and colonization of bacteria in the mucous membrane of the upper respiratory tract, followed by the development of bacterial complications. However, it should be remembered that in some cases other factors may be present and associated, contributing to a decrease in local protective reactions of the mucous membrane and contamination with pathogenic flora, not associated with a respiratory viral infection. These unfavorable factors include: hypothermia, overwork, stress, improper breathing patterns (contributing to the development of dry mucous membranes of the nose, mouth and pharynx), as well as diseases that significantly affect the state of the humoral and cellular immunity, for example diabetes mellitus and its widespread hidden forms (prediabetes - impaired glucose tolerance and impaired fasting glycemia). Thus, it can be assumed that drugs that could affect the severity of symptoms, duration of the disease and the risk of complications are antiviral drugs. Theoretically, this is true, but thanks to the development of evidence-based medicine, it has become known that this conclusion is by no means unambiguous. It should be emphasized here that the mechanism of action of these drugs must be direct antiviral, i.e. the drug must have a proven direct antiviral effect against the influenza virus or any of about 200 different respiratory viruses, for example, blocking its reproduction, penetration into human cells, etc. etc. Let us immediately note that there are very few such drugs. Let's take a closer look at which antiviral drugs are most often prescribed or recommended in our country: these are drugs containing oseltamivir, zanamivir, umifenovir and pentanedioic acid imidazolylethanamide. Oseltamivir and zanamivir
are inhibitors of the neuraminidase enzyme of influenza A and B viruses (this should be especially noted because these substances affect exclusively the influenza virus, but not the other 200 viruses that cause ARVI), due to which the ability of viral particles to penetrate into the cell is impaired.
Global use of both antiviral drugs began in the early 2000s, but the use of oseltamivir, compared with zanamivir, increased sharply after the outbreak of the H1N1 virus (swine or “Mexican” flu) in 2009, despite the same indications and mechanism of action of these two antivirals PM. It was initially thought that the strategy of mass use of neuraminidase inhibitors would lead to a reduction in hospitalizations and complications of influenza, especially serious ones such as pneumonia, during a pandemic. But it turned out that both oseltamivir and zanamivir shorten the duration of flu symptoms by only half a day (from 7 to 6.3 days in adults, and the effect in children is even more insignificant), there is no evidence that they reduce the number of hospitalizations or complications (pneumonia, bronchitis, sinusitis or otitis in adults and children) and reduce the risk of transmission of the virus from one patient to another (as stated at the beginning of their use). This is evidenced by data from a Cochrane review of studies published by the Cochrane Society (an international non-profit organization that studies the effectiveness of medicines and techniques through randomized controlled trials) and the British Medical Journal (BMJ) in April 2014 [3]. The results of trials involving more than 24 thousand people challenged the “historical” glory of neuraminidase inhibitors as an effective tool in the fight against influenza. The evidence obtained in the above studies also suggests that there is no sufficient basis for the use of these drugs in preventing the transmission of the virus from person to person. But important conclusions were made on the tolerability and safety of these antiviral drugs, namely: the use of oseltamivir was associated with such frequent side effects as nausea, vomiting, headache, renal impairment and psychiatric complications (suicidal behavior). The last three occurred when oseltamivir was used to prevent influenza. Oseltamivir's effect on the heart remains unclear: it may reduce heart symptoms, but it may also cause serious heart rhythm problems. In clinical trials of zanamivir in adults, there was no increased risk of reported adverse events. Evidence of possible harm associated with zanamivir treatment in children is mixed. The lack of high-quality evidence demonstrating an effect on influenza complications is consistent with the cautious conclusions for both drugs made by the US Food and Drug Administration (FDA), which described the overall effectiveness of both drugs as “modest” [4]. So, in just a decade in clinical practice, the antiviral drugs oseltamivir and zanamivir have gone from a “panacea” to a “very modest” role in the treatment and recovery of influenza patients. Umifenovir
is another popular antiviral drug in our country (unlike the previous ones, recognized in many countries of the world). According to the mechanism of antiviral action, it belongs to fusion inhibitors, interacts with the hemagglutinin of the virus and prevents the fusion of the lipid membrane of the virus and cell membranes, preventing penetration of the virus into the cell.
Umifenovir has been approved for use in medical practice in adults as a therapeutic and prophylactic agent for influenza A and B since 1988, and in children since 1995. Despite such a long history of use, only in 2013 the working group on the methodology of drug statistics WHO collaborating center assigned the INN umifenovir to umifenovir and included it in the group of direct-acting antiviral drugs of the anatomical-therapeutic-chemical classification in the subgroup “Other antiviral drugs” under the code J05AX13, which does not mean simultaneous recognition of the effectiveness of the drug, as stated in the comments of WHO experts [5]. The effectiveness and safety of umifenovir has been studied in several studies in the post-registration period, mainly conducted in Russia [6]. However, not all clinical studies of umifenovir met quality criteria, especially in terms of recording side effects, which most often were not detected in either the study or control groups, and the conclusion was made that the drug was well tolerated and safe. Information on the profile and frequency of side effects is contained in the reports of only two studies. One of them indicated that the profile and frequency of side effects were similar in the umifenovir and placebo groups, and the most common side effects were gastrointestinal symptoms and increased transaminases [7]. According to the instructions for use, the main side effect of umifenovir is allergic reactions, which are “rare”, which, according to the WHO classification, means the frequency ranges from 1/1000 to 1/10,000. Contraindications for use include hypersensitivity to the drug and age <3 years. Side effects in clinical studies in pediatrics, according to publications, were either not detected at all or nothing was reported about them, although the conclusions contained a statement about the good tolerability and safety of the drug. The instructions for use of the original umifenovir (especially its generics) completely lack any information about the possibility of its use during pregnancy and lactation, however, the “Safety” section on the manufacturer’s website states that “due to the lack of necessary clinical experience, it is not It is recommended to take the drug during pregnancy and breastfeeding, unless, in the physician’s opinion, the possible benefit to the mother outweighs the potential risk to the fetus” [8]. Also, the instructions for use of the drug do not contain any indications of restrictions on its use in the elderly. However, it is known that these patients are at risk for developing complications of influenza and taking umifenovir is strongly recommended for them, and on the other hand, they often suffer from concomitant diseases and take other drugs on an ongoing basis. According to the data, however, there is no information on the study of drug interactions with specific drugs, which, apparently, indicates that drug interactions have not been studied at all. In addition, the “Pharmacokinetics” section does not describe the metabolism of the drug. It is only known that “about 40% is excreted unchanged,” i.e., the remaining 60% of the drug is metabolized. The latter was confirmed in a recent study [9]. According to his results, the main cytochrome P450 isoenzyme involved in the metabolism of umifenovir is CYP3A4. The CYP3A4 enzyme is involved in many fatal drug interactions that have received wide publicity in recent years, which acutely raises the question of the risk of drug-drug interactions when umifenovir is used concomitantly with inducers and inhibitors of this isoenzyme, for example, antibiotics, non-steroidal anti-inflammatory drugs, drugs for the treatment of cardiovascular diseases and endocrinological diseases, etc. [10]. Therefore, the safety of the drug is subject to further study. Until more complete safety data are available, caution should be exercised when using umifenovir, especially in patients at risk for drug-drug interactions. Pentanedioic acid imidazolylethanamide (vitaglutam)
is also a commonly prescribed antiviral drug. The domestic drug vitaglutam for the treatment of influenza was registered in 2008. It should be noted that the active substance itself - imidazolylethanamide of pentanedioic acid (vitaglutam) - for several years, in violation of the Federal Law of April 12, 2010 No. 61-FZ “On the Circulation of Medicines” was included immediately into two drugs from the same manufacturer, produced in different dosage forms under different trade names and recommended for different indications. Initially, pentanedioic acid imidazolylethanamide at a dose of 100 mg was registered to stimulate leukopoiesis in patients receiving chemotherapy with cytotoxic agents for malignancies. And pentanedioic acid imidazolylethanamide in a dose of 90 mg, a drug for the treatment of influenza and other acute respiratory viral infections, was registered later. Despite the same active ingredient of these drugs, different pharmacodynamic data were presented in the instructions for use, which raises doubts regarding its study not only in clinical, but also in preclinical studies. The mechanism of action of the drug vitaglutam as an antiviral drug is also not entirely clear. In cell culture, vitaglutam in non-toxic concentrations (up to 200 μg/ml) did not show antiviral activity against all studied strains of influenza A and B viruses, including strains of the pandemic influenza A (H1N1) virus [11]. In experiments on mice, however, in the same study, vitaglutam had an antiviral effect, but was inferior in effectiveness to umifenovir and rimantadine in influenza pneumonia. This allowed the authors to conclude that the drug does not have a direct virus-specific effect and assume that its effectiveness is due to “other pharmacological properties” [11]. According to the instructions for use of vitaglutam, “the antiviral mechanism of its action is associated with the suppression of virus reproduction at the nuclear phase stage, delaying the migration of the newly synthesized NP virus from the cytoplasm to the nucleus,” however, no publications confirming this could be found in the available literature. The pharmacokinetics of the drug vitaglutam in humans has not been studied, because, according to the manufacturer, its determination in blood plasma at recommended doses is impossible using available methods. The instructions for use contain only data obtained in experiments on animals using a radioactive label, from which it is impossible to reliably judge the pharmacokinetics of the drug in the human body [12–14]. In clinical studies of the drug conducted by the same team of authors, side effects, as a rule, were not observed at all, which once again raises the question of their registration in domestic studies. However, in the English-language abstract of one of the studies, despite the absence of side effects in both the vitaglutam and comparison groups (placebo, umifenovir), it was concluded that vitaglutam was “less toxic” (compared to placebo?) [14, 15]. The drug and its active substance have not been studied in foreign studies. Vitaglutam is contraindicated in persons with individual intolerance to the components of the drug, children and adolescents under 13 years of age and pregnant women. Due to insufficient information about the safety of the drug, its use should be considered with great caution, especially in patients at high risk of developing complications of influenza. As can be seen from the review information presented above, the presence of antiviral activity in a drug (and this activity of many modern drugs that claim this name must still be proven) is not at all synonymous with its effectiveness in the treatment and prevention of influenza and ARVI. A striking example is the sadly discredited neuraminidase inhibitors, which have undergone international research and have been used all over the world for more than 10 years. Moreover, many questions arise regarding drugs that are less studied and do not have such a wide international practice of use, such as umifenovir and vitaglutam, which also do not have proven high efficiency or safety. The next popular group of drugs often prescribed for the treatment and prevention of influenza and ARVI are immunotropic drugs. It should be clarified that under this name it is sold in pharmacies. Numerous so-called immunomodulatory or immunocorrective drugs are most often nonspecific inducers of interferon production or interferon drugs themselves. Interferon inducers are substances of various natures that cause the formation of interferon upon contact with cells and tissues. For example, the most typical and effective inducer of interferon are viruses, but the ability of different viruses to stimulate interferon formation is not the same, i.e., a virus as an inducer of interferon can be strong or weak. Interferon inducers can be not only viruses, but also many bacteria and chemical compounds. It follows that it is possible to stimulate the production of interferon artificially by introducing certain chemicals into the body. This is the basis for the use of this group of drugs. What is the point of these drugs if the virus itself is often a powerful interferonogen? The fact is that different viruses, as we have already written, can have different effects on the production of interferon in the body during infection. Influenza viruses and respiratory viruses are powerful stimulators of the synthesis of interferon protection in the body, so the use of additional stimulants is not justified, as well as additional interferon preparations themselves (neither in the form of drops, nor in the form of suppositories). But the human immunodeficiency virus, herpes virus, cytomegalovirus, as well as viruses that cause hepatitis B and C, are typical examples of viruses that have a weak ability to stimulate the formation of interferons. Therefore, when infected with these viruses, interferon preparations are actively and successfully used. It is important to note here that the predominant way of administering interferon preparations for effective treatment is parenteral [16]. Taking into account the foregoing, it becomes clear why interferon preparations themselves belong to the pharmacological group of drugs that are used to treat mainly viral hepatitis, and are administered exclusively parenterally. Thus, the feasibility of using interferon inducers in the treatment of influenza and acute respiratory viral infections causes quite reasonable doubts, since the vast majority of ARVI pathogens are strong interferon inducers, which means that there is no need for additional stimulation of interferon formation, and in additional interferon drugs. However, such drugs, which have not only low efficiency, but also potential toxicity, are widely prescribed to patients. The most famous is Kagocel (there is no international non-parted name), the chemical name: Sodium Soliper Salt (1 → 4) -6-0-carboximethyl-β-D-Glucosis, (1 → 4) -β-D-Glucose and (21 → 24) -2,3,14,15,21,24, 29,32-octahydroxy-23- (carboxymetoxymethyl) -7, 10-dimethyl-4, 13-d (2- sawdust)-19.22.26, 30.31-pentoropsageptacid [23.3.2.216.20.05.28.08.27.09.18.012.17] Dotriacont-1,3,5 (28), 6.8 (27), 9 (18), 10, 12 (17), 13,15-deecaena. This domestic drug, which causes interferon induction, is registered in 2007 by the acting substance of the drug, according to the manufacturer, is a new chemical substance that is a gossipol (a natural compound contained in the cotton), which is covalently associated with a polymer matrix (oxidized carboxymethyl cellulose). This drug is recommended for adults for the treatment of herpes, for the prevention and treatment of influenza and other SARS, as well as children from 3 years old for the prevention and treatment of influenza and SARS. Thus, it has a wide target audience of patients, including pediatric, in which the incidence of SARS and influenza is the highest [17]. The effectiveness and safety of the drug in adults and children was studied in randomized studies in which its good tolerance was noted. In most studies, there were no undesirable phenomena at all, which actually causes some surprise and doubt about the thoroughness of monitoring, since in well -planned studies, unwanted phenomena are always revealed not only in the main group, but also in the placebo group. The distant consequences of the use of the drug were not studied, although they, in fact, cause serious concern from the point of view of security [18]. The fact is that the stateipol, which is part of the active substance of the Kagocel, is able to inhibit spermatogenesis and was even studied in clinical studies as a male contraceptive [19]. At the same time, in approximately 20% of cases, the effect of the drug on spermatogenesis was irreversible, which involved its use only in men who “completed the formation of families, or those who allow irreversible infertility” [20]. In addition, it is possible that Gossipol has genetic toxicity, because in experimental studies a small, but reliable increase under its influence of the frequency of nursing exchange of chromatids was revealed [21]. In 1998, the WHO research group according to the methods of regulating male fertility, having considered the results of the research of Gossipol, came to the conclusion that the risk of its use exceeds the benefit, and therefore it was prohibited as a contraceptive [22]. The Kagotsel manufacturer claims that the stateipol is not released from the drug due to a strong covalent connection with carboxymethyl cellulose, however, it allows a break in this connection in “special conditions (special chemical reagents, high reaction temperature, etc.)”, which are not found in the human body [23 ]. Meanwhile, the data on the pharmacokinetics of the Kagocel suggests that the release of gossipol can occur in the body. The description of the invention to the patent for the drug indicates that Kagocel is a soluble compound acting at the level of the small intestine [24]. From the instructions for the medical use of the drug it follows that it is relatively well absorbed from the gastrointestinal tract, the bioavailability is 20%, it is widely distributed throughout the body-24 hours after administration, the drug accumulates mainly in the liver, to a lesser extent-in the lungs, timus , spleen, kidneys and lymph nodes. In this case, the high molecular weight compounds, which include the kagocel, for absorption in the gastrointestinal tract, as a rule, need preliminary splitting to smaller molecules. In addition, the instructions for the use of the drug are completely absent from its metabolism. It is noted that 90% of the kagocel is excreted through the intestines and 10% by the kidneys, however, there are no indications that the drug is completely displayed in unchanged form. It should be noted that the possibilities of filtering high molecular weight compounds in the kidneys are limited. The assertion contained in the instructions that Kagocel “does not accumulate in the body” also conflicts with a rather long period of half -life - about 88% of the drug is excreted only a week after the end of its administration. According to the manufacturer, the dose of stateypol in Kagocele is not dangerous for human reproductive health, however, relevant studies were not conducted in the human population either among adults or among children of the prepubertate and puberty. The percentage of the percentage (3%) of the stateypol in the Kagocele, presented on the manufacturer’s website, raises questions from the text of Patent No. 2238122 to the Kagocel drug that the fraction of the gossipol in the Kagotsel molecule is much larger - from 10 to 20% [25]. The information presented on the website of the Kagotsel manufacturer about the doses of stateypol necessary to suppress spermatogenesis is also not quite accurate: “According to the results of a special review analysis of the experts of the European Food Safety Commission, the amount of free gossipol, which must be used to achieve the contraceptive effect, make up 10–20 mg per day, while the achievement of the effect is possible only with a long duration of admission - from 2-3 to 16–18 months ”[26]. At the same time, the publication of EFSA, which is in the public domain on the Internet, indicates that the minimum dose that suppresses spermatogenesis in people is 0.1 mg/ kg [26]. The content of gossipol in the maximum daily dose of a kagocel recommended to persons of different age groups, and the risk of reaching a dose inhibiting spermatogenesis are presented in table 1.
Considering that many children aged 3–6 years weigh <24 kg, those aged 7–11 years weigh <36 kg, and body weight <74 kg is quite common in young men of reproductive age, even with minimal gossypol content in the active substance molecule Kagocel (10%) Many patients may receive an antifertility dose of gossypol. If the Kagocel molecule contains the maximum amount of gossypol described in the patent for the drug (20%), patients of any body weight taking the drug in a therapeutic dosage can receive an antifertility dose. Caution should also be exercised when prescribing Kagocel to women and girls, since there is evidence that gossypol can have an adverse effect on the female reproductive system, and during pregnancy, on the development of the fetus [26]. Thus, the data presented in the instructions for use of Kagocel and on the manufacturer’s website are insufficient, and sometimes incorrect, to assess its safety in patients at risk, and especially to assess the long-term consequences of its use. Until the results of targeted studies are available to study the long-term consequences of using Kagocel (gossypol), especially in males, including children and adolescents, the use of alternative drugs seems safer. To summarize our analysis, we can say that the problems of modern anti-cold therapy are the reduced effectiveness of antiviral drugs, age restrictions, the narrow specificity of a number of drugs, the lack of evidence for a number of drugs and the toxicity of most of them. In addition, when prescribing interferon inducers, it is necessary to take into account that the concentration of interferons in the blood serum of patients with ARVI or influenza at the height of the disease increases significantly, since the virus itself is a powerful interferonogen, and additional artificial stimulation of the production of interferons is fraught with disruption of the compensatory capabilities of the body's immune system [27]. As an alternative to the drugs considered, homeopathic anti-cold drugs for the treatment of acute respiratory viral infections and influenza, which have virtually no contraindications or age restrictions, are free of side effects and can be used in combination with other drugs, are of serious interest [28]. One of the promising homeopathic drugs used in the treatment of ARVI and influenza is Oscillococcinum. Its use is indicated for influenza and colds; a contraindication is increased individual sensitivity to individual components of the drug. The drug is on the market in more than 30 countries around the world, it is especially popular in France, where it has been produced for about 80 years and is the country's best-selling over-the-counter flu medicine. In Russia, the drug Oscillococcinum is registered in accordance with the established procedure and is recommended for use as a drug for the prevention and treatment of mild to moderate influenza and ARVI (registration number P No. 014236/01) [29]. The effectiveness of Oscillococcinum in the treatment of influenza and ARVI has been demonstrated by a number of clinical studies in many countries, including Russia. The results of a multicenter randomized study conducted in France and Germany with the participation of 300 and 372 patients with influenza and influenza-like conditions, respectively, during the epidemiological season of 1990–1991 showed the high effectiveness and safety of Oscillococcinum. All patients who took part in this study were divided into two homogeneous groups - the main and control; in the first, Oscillococcinum was used as an antiviral agent, in the second, a placebo. 2 days after the start of using Oscillococcinum, a significant improvement in condition was noted in 43.7% of patients, there was no deterioration in the condition in any clinical case, and in the placebo group, improvement was observed in 33.5% of patients, deterioration in 5.4%. When using placebo, concomitant therapy (non-steroidal anti-inflammatory drugs, decongestants, mucoactive drugs and a number of others) had to be prescribed 1.5 times more often than in the group of patients taking Oscillococcinum. In addition, in the main group there was a higher percentage of able-bodied patients compared to the control group: 2 days after the start of therapy - 16.3 and 9.2%, after 4 days - 48.9 and 46.7%, respectively [30]. According to the literature, when assessing the clinical use of the drug, a number of studies showed its effectiveness in acute respiratory viral infections of various etiologies in patients of different ages, including early childhood [31]. In a comparative randomized clinical and epidemiological study (epidemiological season 2014–2015), carried out with the participation of 259 children 6–12 years old who were not vaccinated against seasonal influenza, a higher clinical effectiveness of Oscillococcinum was noted in comparison with Kagocel. This applied to both symptoms of intoxication (body temperature, headache) and catarrhal symptoms (hyperemia of the pharynx, sore throat). Patients taking Kagocel had significantly longer manifestations of intoxication syndrome as assessed on the 4th day of treatment (2.1 and 1.82 points for body temperature; 1.18 and 0.06 points for headache; p<0. 05 - for both indicators), until the 7th day they continued to have catarrhal symptoms (1.88 and 0.74 points for hyperemia of the pharynx; 1.6 and 0.38 points for sore throat; p <0.05 – for both indicators) [32]. Thus, the use of Oscillococcinum, according to the results of studies conducted earlier abroad and in our country, provides a rapid reduction in symptoms, shortens the duration of the disease, accelerates recovery and is well tolerated by patients [33–36]. At the same time, Oscillococcinum does not have allergenic or immunotoxic properties [37] and is approved for use in children from the first days of life and in adults with concomitant diseases. Considering the current state of public health, namely the prevalence of chronic diseases in the elderly population, such as bronchial asthma, cardiovascular diseases, kidney and liver diseases, diabetes mellitus, obesity, blood diseases, nervous system diseases, HIV, great attention is needed approach the prescription of anti-cold therapy, giving preference to time-tested, effective and safe drugs.