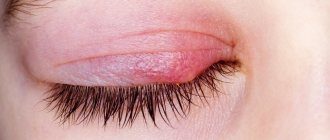
Scleroderma is a systemic autoimmune disease with a high level of risk. In this case we are talking about connective tissue disease. Small vessels and capillaries are affected by the body's own immune cells, as a result of which metabolic processes in tissues deteriorate significantly. The skin, as well as subcutaneous tissue, becomes very dense, as if becoming homogeneous, similar to continuous scar tissue. The disease progresses, affects the joints, internal organs and as a result can lead to the death of the patient.
Most often, scleroderma affects women, and with age, the risks of this pathology increase.
Classification of the disease according to several criteria
If we talk about the clinical form, the classification of scleroderma includes the following options:
- diffuse. It develops quickly and within six months reveals itself with a host of symptoms. Affects the face, head, torso, limbs;
- limited. Mainly affects the face, hands and feet, and develops much more slowly;
- cross. Combined with other autoimmune diseases, such as lupus erythematosus or rheumatoid arthritis;
- visceral (without scleroderma). That is, the skin with this type of disease does not thicken, but Raynaud's syndrome manifests itself (whitening, numbness, blue discoloration of the fingers or toes). The main blow falls on the internal organs - the gastrointestinal tract, lungs, kidneys;
- induced. May be associated with vascular lesions, usually caused by environmental factors, such as exposure to cold or chemicals;
- juvenile This type is typical for early ages, up to 16 years. As a rule, it affects internal organs less, but can cause abnormalities in the development of limbs and other serious problems.
Scleroderma can also be rapidly progressive (acute), moderately progressive (subacute) and slowly progressive (chronic).
SYSTEMIC SCLERODERMA
Systemic scleroderma (SSc) is an autoimmune connective tissue disease, the main manifestations of which are associated with ischemia and fibrosis of organs and tissues. The incidence of SSc is approximately 20 people per 1 million per year. Among the patients, women predominate (the approximate ratio of women to men is 6:1). The peak incidence occurs in the 4th–6th decades of life.
The etiology of the disease is unknown. It is believed that SSc develops under the influence of certain exogenous factors in people with certain genetic disorders. Exogenous factors that can induce the development of SSc include retroviruses (primarily cytomegaloviruses), quartz and coal dust, organic solvents, vinyl chloride, and some drugs (bleomycin and a number of other drugs used for chemotherapy). The pathogenesis of SSc is a combination of many factors, among which immune activation, damage to the vascular endothelium and increased synthetic function of fibroblasts play a key role. The severity of each of these pathogenesis factors varies in individual patients.
As a systemic disease, SSc is characterized by simultaneous damage to the skin, blood vessels, musculoskeletal system and internal organs, including the heart, lungs, kidneys and gastrointestinal tract. At the onset of SSc, before the appearance of specific signs of the disease, constitutional manifestations are often observed: weight loss, low-grade fever, weakness.
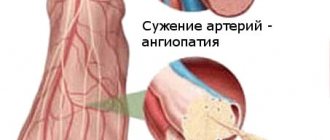
A characteristic early sign of SSc is Raynaud's syndrome (RS) - transient episodes of spasm of the skin vessels of the distal extremities and digital arteries under the influence of cold or emotional stress. Clinically, SR manifests itself as clearly defined areas of discoloration of the fingers. At the beginning of an attack of vasospasm, the fingers become pale in color, which within a few minutes changes to a bluish-violet hue. After the spasm is resolved and blood flow is restored, reactive hyperemia occurs and the skin becomes intensely pink. In some patients, attacks of vasospasm are accompanied by a feeling of freezing of the hands, numbness or paresthesia. During the reactive hyperemia phase, patients may feel pain in the fingers. In the early stages of the disease, these signs can be observed on the distal phalanx of one or more fingers. Subsequently, the affected area spreads to all fingers and, possibly, feet, while the thumbs usually remain intact. Vessels of the skin of the face and other areas may also be subject to vasospasm. In these cases, characteristic changes in the color of the tip of the nose, lips and ears, above the knee joints are observed. In some patients, the vessels of the tongue are also involved in the process, which manifests itself as dysarthria during an attack of vasospasm.
The intensity of SR fluctuates both in different patients and in the same people at different times of the year (more intense in winter than in summer). A three-phase change in skin color (whitening-blue-redness) is not detected in all cases: in some patients, a two-phase or single-phase color change is observed. Depending on the number of phases of skin color change, three-phase, two-phase and single-phase SR are distinguished.
Signs of SR, such as a feeling of freezing of the extremities, numbness and tingling, can be observed in peripheral vascular disease, accompanied by decreased blood flow and ischemia. In SR, unlike peripheral vascular diseases, these symptoms are observed only during vasospasm and disappear completely after restoration of the original blood flow.
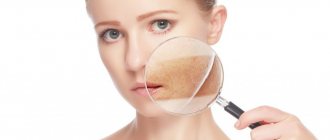
The most specific sign of SSc is skin lesions in the form of thickening and compaction, which are observed in the vast majority of patients with SSc. The severity and prevalence of skin thickening varies among individual patients, but skin thickening in SSc always begins with the fingers and may subsequently spread to the proximal limbs and trunk. Simultaneously with the fingers of the hands, damage to the skin of the face is often observed, as a result of which the nasolabial and frontal folds are smoothed out, the red border of the lips becomes thinner, around which radial wrinkles appear, and the oral aperture decreases (pouch-purse symptom). With long-term observation, the stages of skin damage are noted: swelling, induration, atrophy. Skin thickening tends to progress over the first 3–5 years of the disease. In later stages of the disease, the skin becomes less dense and the compaction remains only on the fingers.
Often a sign of SSc is hyperpigmentation, limited or diffuse, with areas of hypo- or depigmentation (“salt and pepper”). A characteristic symptom is ischemic digital ulcers (so named because of their typical localization on the distal phalanges of the hands), which can be sharply painful, characterized by torpidity during treatment and a recurrent course. Ulcerative skin lesions are also observed in other areas exposed to mechanical stress: above the elbow and knee joints, in the ankles and heels. As a result of ischemic disorders, digital scars and pinpoint areas of skin atrophy (“rat bite”) appear. Digital scars can also appear after digital ulcers have healed. Due to atrophy of the hair follicles, sweat and sebaceous glands, the skin in places of compaction becomes dry and rough, and loses hair. Telangiectasia with a characteristic localization on the fingers and face, including the lips, is a late sign of the disease. Small subcutaneous calcifications usually appear in the later stages of the disease in areas that are often subject to microtrauma. Calcifications are usually painless, but can cause local inflammation and rupture with the release of a curdled mass.
Arthralgia and morning stiffness are common manifestations of SSc, especially in the early stages of the disease, but arthritis is detected in a small number of patients. Due to compaction of the skin of the fingers, flexion contractures of small joints of the hands develop, and with widespread compaction of the skin, large joints. Sometimes polyarthritis can resemble joint damage in rheumatoid arthritis (RA), but unlike the latter, it is characterized by a predominance of fibrous periarticular changes. Tenosynovitis can lead to carpal tunnel syndrome and a peculiar symptom of friction of the tendons of the distal forearms, determined by palpation during active movements of the hands. The result of ischemia is osteolysis of the nail phalanges, manifested by shortening and deformation of the fingers. In some cases, osteolysis of the distal parts of the radius and ulna and the processes of the branches of the lower jaw is observed.
Muscle damage can lead to the development of clinical manifestations of inflammatory myopathy (proximal muscle weakness, increased creatine kinase, characteristic changes in electromyography and muscle biopsies). The more common form of muscle damage in SSc is non-inflammatory, non-progressive fibrous myopathy.
Damage to the gastrointestinal tract (GIT) develops in 90% of patients with SSc and is clinically manifest in half of them. Dysfunction of the distal esophagus, the most common manifestation of gastrointestinal lesions, is observed in 80–90% of patients and is often one of the first symptoms of the disease. Involvement of the esophagus is manifested by dysphagia, persistent heartburn, which worsens after eating. Dysphagia can be a consequence of both hypotension and esophageal stricture. The most sensitive method for detecting esophageal hypotension is manometry. In SSD, there is a decrease in the amplitude of peristaltic waves and the pressure of the lower esophageal sphincter. Hypotension of the esophagus is manifested by expansion of the lumen and an increase in the passage time of barium mass through the esophagus during x-ray examination. Chronic esophagitis is often complicated by erosive lesions of the esophageal mucosa. Endoscopic examination may reveal Barrett's metaplasia. Slowing the evacuation of food from the stomach also aggravates the symptoms of reflux, often causing nausea and vomiting. Damage to the stomach and duodenum is manifested by abdominal pain and flatulence. Damage to the small intestine is often asymptomatic, but with pronounced changes, malabsorption syndrome develops with diarrhea, flatulence and weight loss, and pseudo-obstruction phenomena also occur. The consequence of damage to the large intestine is constipation and incompetence of the anal sphincter.
Lung damage develops in more than 70% of patients with SSc and manifests itself in two clinical and morphological variants: interstitial pulmonary fibrosis and pulmonary hypertension (primary or secondary). Pulmonary fibrosis develops in the early stages of SSc in most patients and is usually limited to the basal regions (basal pulmonary fibrosis). In some patients, pulmonary fibrosis is widespread, leading to a significant decrease in lung volumes, the development of severe respiratory failure and fibrosing alveolitis. Both pulmonary fibrosis and pulmonary hypertension are manifested by increasing shortness of breath and persistent nonproductive cough. A highly sensitive method for detecting pulmonary fibrosis is high-resolution computed tomography. At the early, exudative stage of pulmonary fibrosis, changes of the so-called ground glass type are determined, and at the late fibrous stage, changes of the reticular type are detected. X-ray examination reveals changes in the pulmonary pattern due to interstitial fibrous changes in the basal and parapleural parts of the lungs. A study of the function of external respiration shows an isolated decrease in the forced vital capacity of the lungs, i.e., a restrictive type of disorder, which is accompanied by a decrease in the diffusion capacity of the lungs due to thickening of the interalveolar septa. A characteristic auscultatory phenomenon in pulmonary fibrosis is crepitation, heard at the height of inspiration and reminiscent of the crunching of cellophane.
Pulmonary hypertension is detected in approximately 10% of patients and can be primary or secondary. Primary pulmonary hypertension develops in the later stages of the disease (after 10–15 years) without clinical and instrumental signs of severe pulmonary fibrosis. Secondary pulmonary hypertension is associated with severe pulmonary fibrosis, appears in the first years of the disease and differs in its genesis from primary hypertension. The only complaint of patients is shortness of breath, the severity of which correlates with the degree of increase in pressure in the pulmonary artery. However, in approximately 1/3 of patients, pulmonary hypertension is asymptomatic, especially in the early stages. The screening method for detecting pulmonary hypertension is echocardiography. A reliable way to diagnose pulmonary hypertension is to catheterize the right side of the heart and measure the pressure in the pulmonary artery. The presence of pulmonary hypertension is indicated by a decrease in the diffusion capacity of the lungs with an unchanged forced vital capacity of the lungs, i.e. in the absence of restrictive disorders. The ECG reveals signs of overload of the right heart. Chest x-ray shows dilation of the pulmonary artery and weakening of the vascular component of the pulmonary pattern. In rare cases of the development of pleurisy in patients with SSc, pain is observed when breathing, and sometimes a pleural friction noise is heard. Currently, lung damage is the main cause of death in SSc.
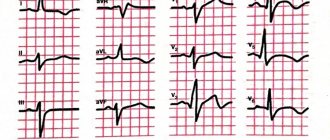
Clinical signs of heart damage in the form of left ventricular dysfunction, conduction and rhythm disturbances, adhesive or exudative pericarditis are detected in the majority of patients during a targeted study. In approximately 10% of patients, the ECG reveals focal myocardial fibrosis, which is not associated with coronary disease and is a consequence of vasospasm of small vessels (the so-called visceral Raynaud's syndrome). The ECG can also detect signs of focal ischemia, which are persistent and often occur without clinical symptoms. Heart lesions are manifested by complaints of discomfort or prolonged dull pain in the precordial region, palpitations and arrhythmias. Signs of myocarditis are observed almost exclusively in patients with symptoms of polymyositis. Heart failure develops rarely, is refractory to therapy and has a poor prognosis. Rare manifestations of heart damage include endocarditis with the formation of heart defects. Along with lung damage, heart damage occupies a significant place in the structure of mortality in patients with SSc.
Kidney disease in the form of acute scleroderma kidney in the European population occurs in 4-5% of patients. Characteristic manifestations of scleroderma kidney are the sudden development and rapid progression of oliguric renal failure, malignant hypertension with high renin levels, thrombocytopenia and microangiopathic hemolytic anemia. This type of lesion usually develops in the first five years of the disease. Latent nephropathy, manifested by impaired renal concentration function, moderate proteinuria and decreased renal functional reserve, is more common. Morphological examination reveals kidney damage in more than 80% of patients and is characterized primarily by changes in the renal vessels. In approximately 10% of patients, renal crisis occurs in the presence of normal blood pressure. Recently, cases of normotensive nephropathy associated with antineutrophil cytoplasmic antibodies induced by D-penicillamine have appeared. Despite certain advances in the treatment of this disease, acute scleroderma kidney remains a potentially fatal complication of SSc, characterized by high mortality (up to 50% during the first year).
Damage to the nervous system manifests itself in patients with SSc predominantly as polyneuritic syndrome, which may be associated with Raynaud's phenomenon or primary damage to peripheral nerves. Trigeminal sensory neuropathy is observed in 10% of patients, which manifests itself as unilateral or bilateral facial numbness, often in combination with pain or paresthesia. In rare cases, damage to the facial, glossopharyngeal or auditory nerves occurs. With severe thickening of the skin of the forearms, carpal tunnel syndrome often develops.
Other common manifestations of SSc include Sjogren's syndrome (20%) and thyroid disease (Hashimoto's thyroiditis or De Quervain's thyroiditis), leading to the development of hypothyroidism.
The basis for the classification of SSc is the prevalence of skin lesions, which correlates with the course of the disease and the nature of the visceral pathology.
According to this classification, there are two main clinical forms of SSc: limited and diffuse. The limited form is characterized by the following symptoms: Raynaud's syndrome precedes the appearance of other signs of the disease for many years; skin lesions are limited to the face and distal extremities; late development of pulmonary hypertension with/without interstitial pulmonary fibrosis; high frequency of detection of anticentromere antibodies (in 70–80% of patients); capillary dilatation without significant avascular areas.
The diffuse form has its own characteristics: the development of skin changes during the first year after the onset of Raynaud's syndrome; involvement of the skin of all parts of the limbs and trunk; palpation detection of tendon friction; early development of interstitial fibrosis of the lungs, damage to the gastrointestinal tract, kidneys and myocardium; expansion and reduction of capillaries; antibodies to topoisomerase-1 (Scl-70) and RNA polymerases.
In both forms, thickening of the facial skin is observed. Usually, during the first year of the disease, the type of skin lesion can be determined. Clinical observations have shown that the course of the disease, the frequency and severity of visceral lesions in SSc correlate with the prevalence of skin lesions.
Diffuse skin damage is accompanied by a progressive course with early and significant damage to internal organs during the first five years of the disease and pronounced constitutional manifestations. The limited form of SSc, on the contrary, is characterized by slow progression with late development of visceral lesions.
In some cases, with obvious signs of visceral lesions specific to SSD, there is no thickening of the skin, i.e., so-called scleroderma without scleroderma is observed. This form of the disease is characterized by: clinical signs of pulmonary fibrosis, damage to the kidneys, heart and gastrointestinal tract; no skin lesions; possibility of Raynaud's syndrome; possible detection of antinuclear antibodies to topoisomerase-1 (Scl-70) and RNA polymerases.
Some authors identify prescleroderma as a special clinical form, diagnosed on the basis of capillaroscopic changes, detection of autoantibodies to topoisomerase-1, centromeric proteins and RNA polymerases, in patients with isolated Raynaud's syndrome.
Clinical signs of SSc are possible in combination with signs of other autoimmune rheumatic diseases (systemic lupus erythematosus, rheumatoid arthritis, dermatomyositis, vasculitis).
Conventional laboratory parameters are uninformative and their changes are nonspecific for SSc. In approximately half of the cases, an increase in ESR of more than 20 mm/h is observed. With the same frequency, signs of inflammatory activity are detected in SSc: increased levels of fibrinogen and seromucoid; Rarely, an increase in C-reactive protein levels is observed.
In 10–20% of patients, anemia is detected, the cause of which may be iron and vitamin B12 deficiency, kidney damage (microangiopathic hemolytic anemia) or bone marrow itself (hypoplastic anemia). The identification of SSc-specific autoantibodies is of great importance.
Among the many instrumental research methods used for early and differential diagnosis of SSc, it is difficult to overestimate the role of capillaroscopy of the nail bed. Characteristic structural changes in the form of dilatation and reduction of capillaries are detected in the initial stages of the disease, before the development of the main clinical signs, which makes it possible to clearly differentiate SSc from many other systemic connective tissue diseases. Methods for studying microcirculation, such as laser Doppler flowmetry, plethysmography and others, are of secondary importance in the diagnosis of SSc due to the significant variability of results.
Treatment of SSD
Therapy is always prescribed individually, depending on the clinical form and course of the disease, the nature and degree of ischemic and visceral lesions. Before starting treatment, the patient should be convinced of the need for long-term therapy, strict adherence to recommendations, and familiarized with the possible side effects of the medications used. Considering the progressive course of the disease in most cases, it is important to draw the patient’s attention to the need for constant medical supervision and regular examinations for early detection of signs of disease progression and possible correction of therapy.
Therapy is carried out for the purpose of: prevention and treatment of vascular complications; suppressing the progression of fibrosis of the skin and internal organs; effects on the immunoinflammatory mechanisms of SSc; prevention and treatment of damage to internal organs.
Patients need to reduce their time in the sun, avoid prolonged exposure to cold, and local exposure to vibration. To reduce the frequency and intensity of vasospasm attacks, it is recommended to wear warm clothing, including heat-retaining underwear, hats, woolen socks and mittens (instead of gloves). For the same purpose, the patient is advised to stop smoking and stop drinking coffee and caffeine-containing drinks.
The main directions of drug treatment are vascular, antifibrotic and immunosuppressive therapy. Vascular therapy is carried out to reduce the frequency and intensity of episodes of vasospasm (Raynaud's syndrome) and improve blood fluidity and includes the use of vasodilators, as well as drugs that affect blood viscosity and platelet aggregation. The most effective vasodilators are calcium channel blockers. According to their chemical structure, they are divided into four main groups: phenylalkylamines (verapamil, gallopamil), dihydropyridines (nifedipine, amlodipine, nicardipine, isradipine, lacidipine, nimodipine, nitrendipine, riodipine, felodipine, etc.), benzothiazepines (diltiazem, etc.) and derivatives piperazine (cinnarizine, flunarizine).
Among all groups of calcium channel blockers, dihydropyridine derivatives exhibit the greatest selectivity for vascular smooth muscle cells and, accordingly, a vasodilatory effect. The drug of choice is nifedipine (calcigard retard, cordafen, cordipine, nifedex, nifecard), the effective daily dose of which is 30–60 mg in three or four doses. Nifedipine significantly reduces the frequency and intensity, and in some cases, the duration of episodes of vasospasm. The effectiveness of nifedipine varies in individual patients and is less pronounced in secondary Raynaud's syndrome compared to patients with primary Raynaud's syndrome. Approximately 1/3 of patients treated with nifedipine develop side effects characteristic of most dihydropyridine derivatives, among which the most common are reflex tachycardia, headache, dizziness, facial flushing and swelling of the legs (pretibial myxedema). The development of side effects is associated with systemic arterial hypotension and the negative chronotropic effect of the drug. Recently, retard forms of nifedipine (calcigard retard, cordipine retard) have been increasingly used, which create a relatively constant concentration of the drug in the blood and thereby reduce fluctuations in blood pressure and associated side effects.
If nifedipine is intolerant, other dihydropyridine derivatives may be prescribed. Amlodipine (amlovas, kalchek, norvasc, normodipine) has a prolonged effect and is prescribed once in a dose of 5–10 mg. Amlodipine significantly reduces the frequency and severity of vasospastic attacks, and also eliminates changes in digital blood flow associated with post-ischemic reactive hyperemia. The most common side effect of amlodipine is ankle swelling, which occurs in approximately 50% of patients. Isradipine (Lomir) is prescribed in a daily dose of 5 mg in two doses. If the effect is insufficient and is well tolerated, the daily dose can be increased to 10 mg. The most common complications of isradipine treatment are headache and facial flushing. Felodipine (auronal, plendil, felodil) in a daily dose of 10–20 mg reduces the frequency and severity of vasospasm to a degree comparable to the effect of nifedipine.
Diltiazem (altiazem RR, diazem, diltazem SR) at a therapeutic dose of 180 mg/day is less effective than nifedipine, but is better tolerated. Diltiazem has no significant effect on blood pressure and pulse at their normal baseline values and reduces tachycardia. If you take a larger dose, swelling of the ankles and headache may occur. Verapamil does not have a vasodilator effect. With long-term use of calcium channel blockers, the possibility of developing refractoriness must be taken into account.
If there are contraindications or intolerance to calcium channel blockers, vasoactive drugs of other groups are used. Pathogenetically justified is the prescription of α2-adrenergic receptor blockers (dihydroergotamine, doxazosin, nicergoline, prazosin, terazosin). Good results are observed with treatment with a standardized extract of ginkgo biloba (tanakan - tablets 40 mg 3 times a day). In particularly severe cases (for example, pulmonary hypertension, renal crisis, gangrene), synthetic prostaglandin E1 (alprostadil) is used at a dose of 20-40 mcg intravenously for 15-20 days or prostacyclin analogues (iloprost).
The effectiveness of treatment of vascular manifestations of SSc increases with the inclusion in therapy of antiplatelet agents (acetylsalicylic acid, ginkgo biloba, dipyridamole, pentoxifylline, ticlopidine) and, if necessary, anticoagulants (acenocoumarol, warfarin, heparin sodium, dalteparin sodium, nadroparin calcium, enoxaparin sodium, ethyl biscoumacetate). The combination of vasodilators and antiplatelet agents makes it possible to prescribe the minimum effective dose of each of these drugs and thereby reduce the incidence of side effects. For this purpose, pentoxifylline is most widely used in a daily dose of 600–1200 mg. Pentoxifylline improves the rheological properties of blood by reducing platelet and erythrocyte aggregation, and also has a vasodilatory effect by blocking phosphodiesterase. In cases of multiple ulcerative lesions that are refractory to conventional treatment, a short course (10–15 days) of anticoagulant therapy, preferably with low molecular weight heparin, is indicated.
Antifibrotic therapy is prescribed for the diffuse form of SSc. D-penicillamine, the main drug that suppresses the development of fibrosis, disrupts collagen synthesis by cleaving cross-links between newly synthesized tropocollagen molecules. Penicillamine (artamine, cuprenil) affects various parts of the immune system (selective inhibition of activity and suppression of the synthesis of interleukin-2 by CD4 + T lymphocytes), and has an antiproliferative effect on fibroblasts. The effective dose of the drug is 250–500 mg/day. Penicillamine is taken exclusively on an empty stomach. The previously practiced use of high doses of the drug (750–1000 mg/day) does not significantly increase the effectiveness of therapy, but much more often causes complications, due to which it is necessary to interrupt treatment. If side effects develop (dyspepsia, proteinuria, hypersensitivity, leukopenia, thrombocytopenia, autoimmune reactions, etc.), it is necessary to reduce the dose or discontinue the drug. The basis for discontinuation of penicillamine is proteinuria above 2 g/day. Due to the high frequency of side effects (up to 25%), which are often dose-dependent, during treatment it is necessary to carefully monitor patients, do blood and urine tests every 2 weeks in the first 6 months of treatment, and then once a month .
Anti-inflammatory (immunosuppressive) therapy . NSAIDs (diclofenac, ibuprofen, ketoprofen, meloxicam, nimesulide, piroxicam, celoxib) in standard therapeutic doses are indicated for the treatment of muscular and joint manifestations of SSc, persistent low-grade fever (high fever is not typical for SSD). Glucocorticoids (betamethasone, hydrocortisone, dexamethasone, methylprednisolone, prednisolone, triamcinolone - no more than 15–20 mg/day) are prescribed for obvious clinical signs of inflammatory activity (myositis, alveolitis, serositis, refractory arthritis, tenosynovitis) and in the early (edematous) stage of SSc. , but do not affect the progression of fibrosis. Taking higher doses increases the risk of developing normotensive renal crisis.
Symptomatic therapy . If the esophagus is affected, frequent split meals are recommended. To relieve dysphagia, prokinetics are prescribed in short courses: domperidone, meclozine, ondansetron, metoclopramide; for reflux esophagitis - proton pump inhibitors (omeprazole 20 mg/day, lansoprazole 30 mg/day, rabeprozole, etc.). Long-term use of metoclopramide is unacceptable, since it is associated with the development of neurological disorders (parkinsonism) caused by effects on dopaminergic structures of the brain. If a hiatal hernia develops, surgical treatment is indicated.
When the small intestine is damaged, antibacterial drugs are used: erythromycin (sineryt, erythromycin, erifluid), ciprofloxacin (quintor, siflox, ciprovin, cipromed, ciprofloxacin), amoxicillin (ranoxil, flemoxin solutab, hiconcil), metronidazole (metronidazole, trichopolum). Antibiotics must be changed every 4 weeks to avoid refractoriness. In the early stage, prokinetics are prescribed; if pseudo-obstruction develops, the synthetic somatostatin analogue octreotide (100–250 mg 3 times a day subcutaneously) is recommended.
For interstitial pulmonary fibrosis, low doses of prednisolone and cyclophosphamide are prescribed. The effectiveness of penicillamine in interstitial pulmonary fibrosis has not been proven. A good effect is observed in most cases with intravenous pulse therapy with cyclophosphamide at a dose of 1 g/m2/month in combination with prednisolone at a dose of 10–20 mg per day. The effectiveness of therapy is evidenced by the stabilization of the forced vital capacity of the lungs, since improvement in the function of external respiration at the stage of reticular changes in the lungs is unlikely. Pulse therapy with cyclophosphamide continues at the indicated dose for at least 6 months (in the absence of side effects). If the dynamics of pulmonary function tests and radiological changes are positive, the interval between pulse therapy with cyclophosphamide increases to 2 months, and if the positive dynamics are maintained - 3 months. Pulse therapy with cyclophosphamide must be continued for at least 2 years. The use of these drugs not only slows down the progression of pulmonary fibrosis, but also has a positive effect on the manifestations of pulmonary hypertension.
Treatment of pulmonary hypertension traditionally includes the use of vasodilators (calcium channel blockers) and indirect anticoagulants (acenocoumarol, warfarin) in therapeutic doses. Calcium channel blockers (nifedipine) are indicated only when a decrease in pulmonary artery pressure is detected by right heart catheterization after taking a single dose of the drug. Targeted studies have shown that nifedipine reduces pulmonary artery pressure in only 25% of patients with SSc complicated by pulmonary hypertension. Great success has been achieved in the treatment of pulmonary hypertension after the use of epoprostenol (prostacyclin) and the non-selective blocker of endothelin-1 receptors type A and B bosentan.
Treatment of heart failure is carried out according to generally accepted regimens. Particular attention should be paid to the inadmissibility of excessive diuresis, leading to a decrease in the effective plasma volume and provocation of a renal crisis.
For scleroderma renal crisis, the drugs of choice are the ACE inhibitors captopril (capoten, captopril) - at a dose of 12.5–50 mg 3 times a day, enalapril (berlipril 5, invoril, renipril, enarenal, envas) - 10–40 mg/day, which should be prescribed as early as possible (preferably within the first 3 days) from the moment the first signs of kidney damage appear. Calcium channel blockers are less effective. In case of progression of renal failure, hemodialysis is necessary. It should be emphasized that plasmapheresis, the prescription of glucocorticoids and cytotoxic drugs are contraindicated, since they do not affect the progression of renal pathology, but, on the contrary, can contribute to an exacerbation of the process.
SSD and pregnancy . Most patients with SSc have a history of one or more pregnancies and births. The limited form and chronic course of SSc are not a contraindication for pregnancy. However, during pregnancy, the development of organ pathology may occur, which requires regular examination of their functional state. Contraindications to pregnancy in SSc are a diffuse form of the disease, severe dysfunction of internal organs (heart, lungs and kidneys). In cases of SSc detection during pregnancy, careful monitoring of renal and cardiac functions is necessary.
The prognosis for SSc remains the most unfavorable among systemic connective tissue diseases and largely depends on the clinical form and course of the disease. According to the results of a meta-analysis of 11 studies, the 5-year survival rate of patients with SSc ranges from 34 to 73% and averages 68%. Predictors of an unfavorable prognosis are: diffuse form; age of onset of disease over 45 years; male gender; pulmonary fibrosis, pulmonary hypertension, arrhythmia and kidney damage in the first 3 years of the disease; anemia, high ESR, proteinuria at the onset of the disease.
All patients with SSc are subject to clinical observation in order to assess the current activity of the disease, for the timely detection of organ pathology and when indicated for correction of therapy. A medical examination is carried out every 3–6 months depending on the course of the disease, the presence and severity of visceral lesions. At the same time, general and biochemical blood and urine tests are performed. During repeated visits to the doctor, it is necessary to actively question the patient in order to assess the dynamics of Raynaud's syndrome, increased manifestations of esophageal reflux, shortness of breath, cardiac arrhythmia, etc. When examining the patient, you should pay attention to the prevalence and severity of skin thickening, basal crepitus of the lungs, and increased blood pressure , the presence of digital ulcers and edema. Pulmonary function testing and echocardiography are recommended. In patients taking warfarin, the prothrombin index and international normalized ratio should be monitored, and when treated with cyclophosphamide, general blood and urine tests should be examined once every 1–3 months.
R. T. Alekperov , Candidate of Medical Sciences, Institute of Rheumatology, Russian Academy of Medical Sciences, Moscow
Symptoms
Since the disease comes in different types and has a lot of features, there is also no clear picture with the symptoms. For this reason, it is customary to highlight general trends, and then, in the nuances, doctors understand the diagnostic process.
So, the general signs of scleroderma are:
- changes in the skin, which are characterized either by Raynaud's syndrome or its presence and compaction of individual areas like scar tissue. Moreover, the size of these seals can be quite impressive. If scleroderma develops on the face, it can give the so-called mask effect - when facial expressions are difficult, and the skin looks lifeless, doll-like;
- vascular disorders. Raynaud's syndrome also applies here, but since it also gives a visual effect (white, red or already bluish fingers), we included it in the item associated with skin changes;
- disorders of the musculoskeletal system due to damage to connective tissue.
- a variety of changes in internal organs - from subtle to fatal.
Obviously, not all of these symptoms of scleroderma can be seen and detected by people themselves. If visually the disease proceeds without any significant changes, then the patient’s condition will indicate the danger. Weakness, fatigue, temperature up to 38 degrees are also signs indicating that it is time to go to the doctor and get diagnosed.
Since the pathogenesis of scleroderma often involves the involvement of mucous tissues in the process, there are signs that are specific to these areas. For example, patients develop rhinitis, stomatitis and other similar problems.
As for bones and joints, a person may experience stiffness of movement, the inability to quickly bend and straighten a limb, as well as swelling. It gets to the point that in some areas small bones can even dissolve, causing curvature and other unpleasant consequences. Due to impaired joint mobility, some muscles may atrophy or significantly weaken.
Are you experiencing symptoms of scleroderma?
Only a doctor can accurately diagnose the disease. Don't delay your consultation - call
Signs related to internal organs
Since the disease involves connective tissue and greatly affects internal organs, patients can have a full range of problems with the gastrointestinal tract, heart, kidneys and other systems. They are manifested by any signs of the corresponding diseases. Very often (in 70% of cases) the lungs are affected - it is lung diseases that often cause the death of the patient. In this case, scleroderma can cause pneumonia, abscesses and even lung cancer, which is 3-5 times more common in people with this disease than in other representatives of the same age groups.
It is important to understand that there is no clear list of symptoms in this case - otherwise it would be huge. The disease is dangerous because it affects almost the entire body and can manifest itself in any way.
Etiology
Perhaps the disease has a genetic predisposition. However, reliably provoking factors for its occurrence are external harmful factors such as hypothermia, vibration at work, and previous infections of the nervous system. The development of inflammation of small vessels leads to the growth of collagen and fibrous tissue around them, as well as a specific change in their walls - thickening, loss of elasticity, and possibly even complete closure of the lumen of small vessels. These changes, in turn, lead to disruption of the blood supply to all organs and tissues involved in the pathological process. Insufficient blood supply to tissues leads to their thinning (for example, the mucous membranes of the esophagus and stomach), or, conversely, thickening (the walls of the alveoli in the lungs), disruption of their basic functions (absorption in the gastrointestinal tract, removal of carbon dioxide by the lungs, contraction of muscle fibers).
Diagnostics
To diagnose scleroderma, a set of methods is used:
- blood tests, including biochemical;
- X-ray of feet and hands, CT, MRI, as well as ultrasound of other organs;
- functional tests. These include tests to determine exercise tolerance and tests for respiratory dysfunction;
- examination of small skin samples under a microscope.
An integrated approach and a thorough visual examination of the patient help make the diagnosis as accurately as possible.
Treatment
This autoimmune pathology has not yet been eliminated - such a cure does not currently exist. All actions are aimed at stabilizing the patient’s condition and prolonging his life. Treatment of scleroderma for this purpose includes:
- taking various medications: antifibrotic, anti-inflammatory drugs, enzymes, etc.;
- surgical intervention. It is necessary to restore the mobility of the limbs, to eliminate Raynaud's syndrome, as well as to correct cosmetic imperfections (mainly on the face);
- physiotherapy. This includes laser blood irradiation (low-intensity), as well as PUVA therapy sessions;
- nutrition correction. A diet for scleroderma involves choosing healthy foods that are as rich as possible in vitamins and microelements.
Patients suffering from this pathology are advised to give up bad habits and move towards a healthy lifestyle.
Forecast
Life expectancy and prognosis for scleroderma are determined by the nature of its course. In accordance with this, the following types of disease are distinguished:
- Spicy. Scleroderma is characterized by rapid development and early onset of severe clinical symptoms. In this case, the prognosis for recovery and life is considered unfavorable. As a rule, the acute course of the pathology leads to multiple organ damage a year after the onset of the disease. Acute scleroderma is considered the most dangerous and severe form of the disease.
- Subacute. The autoimmune disease develops quickly, however, tissue sclerosis occurs more slowly than in the acute course. This provides a more favorable prognosis. The success of therapy depends on the degree of activity of scleroderma.
- Chronic. The development of the disease occurs slowly. In this regard, the prognosis becomes favorable.
Scleroderma is an incurable disease. Compliance with treatment and lifestyle recommendations allows you to maintain long-term remission. Preventive measures include:
- Regular medical examination. This is necessary when signs of exacerbation appear, as well as in the case of a stable condition to control the disease over time.
- Taking medications. Self-adjustment of the dose of prescribed medications is not allowed without consultation with your doctor.
- Avoiding hypothermia and fatigue. A prolonged stressful situation can provoke an exacerbation of scleroderma. Therefore, it is necessary to observe a work schedule with rest breaks.
- Maintaining a rational and balanced diet. There is no specific diet for scleroderma. However, it is recommended to include foods rich in vitamins, minerals, and microelements in your daily diet. The menu limits the amount of salt and water, as well as vitamin C due to its stimulating effect on the growth of connective tissue.
Compliance with the above recommendations will help minimize the number of exacerbations of scleroderma. When the first signs of the disease appear, you must seek medical help for diagnostic measures. In this way, the development of complications and severe scleroderma can be prevented.