Symptoms
Signs of the disease are different, they depend on where exactly the amyloid deposits are localized, how widespread the disease is, and whether there are complications. Often there is a complex of symptoms reflecting damage to several organs.
With amyloidosis of the gastrointestinal tract the following are observed:
- tongue enlargement;
- difficulty swallowing;
- bowel dysfunction;
- heartburn, nausea;
- stomach ache.
Signs of liver amyloidosis:
- change in liver size;
- pain in the hypochondrium on the right;
- nausea, belching;
- jaundice.
Pancreatic amyloidosis is characterized by dull pain in the left hypochondrium.
Cardiac amyloidosis is expressed in rhythm disturbances, myocardial lesions, and heart failure.
Amyloidosis of the nervous system has the following symptoms:
- peripheral polyneuropathy (numbness of the limbs, tingling, burning sensation);
- headaches, dizziness, increased sweating;
- urinary and fecal incontinence;
- sexual dysfunction.
With amyloidosis of the respiratory system, hoarseness of the voice and bronchitis are observed.
Classification and epidemiology
In AS, the myocardium is infiltrated with an insoluble glycoprotein, amyloid, formed from various serum or locally produced precursor proteins. These include constant proteins that are independent of amyloid type, such as serum amyloid P protein (SAP), but about 80% of the amyloid mass consists of proteins that differ between amyloid types. Modern classification of amyloidosis is based on differences in amyloidogenic precursor proteins. About 30 such proteins are currently known; Accordingly, there are about 30 forms of amyloidosis. Each form is designated by an abbreviation that includes the designation of the precursor protein: AL (L - light chains of immunoglobulins), ATTR (TTR - transthyretin), CAA (serum amyloid A), etc. (see table). According to this classification, primary and myeloma-associated amyloidosis should be abbreviated AL, since amyloid in these forms is formed by light chains of immunoglobulins. In systemic forms of amyloidosis (most cases of AL, as well as ATTR, AA, etc.), the amyloidogenic precursor protein circulates in the blood and can be deposited in various tissues, the tropism for which differs among different proteins. In addition, local amyloidosis, for example in the atria, may develop due to local production of the amyloidogenic variant of atrial natriuretic factor (AANF).
. Classification of cardiac amyloidosis.
The most common variant of systemic amyloidosis is AL amyloidosis, one of the many forms of plasma cell dyscrasia. The term “plasma cell dyscrasia” unites a group of conditions caused by pathology of plasma cells in the bone marrow, and less commonly, other tissues. In this case, in the bone marrow there is a dominance of the pathological clone of plasma cells that synthesize abnormal immunoglobulins or their components: heavy or light chains. In addition to AL amyloidosis, plasma cell dyscrasias include multiple myeloma, Waldenström macroglobulinemia, etc.
In AL amyloidosis, a pathological clone of plasma cells synthesizes light chains of immunoglobulins, the deposition of which in tissues is accompanied by the formation of insoluble amyloid fibrils. AL amyloidosis is the most severe, generalized form of the disease, affecting many tissues. The heart is affected in 90% of patients, half of whom develop diastolic heart failure (HF) [2]. Death usually occurs from heart failure or arrhythmias [4].
The isolated cardiopathic variant of AL amyloidosis is rarely diagnosed - in less than 5% of patients [5]. Isolated cardiac damage is characteristic of AANF amyloidosis, in which infiltration of the atria with amyloid formed from atrial natriuretic peptide is noted. This form of amyloidosis is found in more than 95% of people over 80 years of age [6]. AANF amyloidosis rarely leads to HF [2], but can cause the development of supraventricular arrhythmias [7–9].
Heart damage is typical for acquired and various variants of hereditary ATTR amyloidosis. This form is caused by the deposition of the protein that transports thyroid hormones and retinol - transthyretin. It is secreted by the liver in a tetrameric form consisting of four transthyretin molecules. With age, a decrease in the activity of hepatocyte enzyme systems leads to an increase in the secretion of amyloidogenic monomeric forms of transthyretin and, as a result, the development of amyloidosis. Acquired ATTR amyloidosis is usually called systemic senile amyloidosis (affects up to 25–36% of people over 80 years of age). Amyloid is found in many organs, but the most significant deposits are found in the heart. Typically, the disease has a benign, asymptomatic course, manifested by blockade of the anterior branch of the left bundle branch and slight thickening of the walls of the left ventricle (LV). With significant amyloid deposition, cardiomegaly and slowly progressive heart failure may develop [3].
In the presence of mutations in the transthyretin molecule, the assembly of tetramers in the hepatocyte is initially disrupted, which leads to the development of hereditary ATTR amyloidosis at an earlier age. Currently, about 100 point mutations and deletions of the transthyretin gene are known, 87 of which are amyloidogenic [10]. The disease is characterized by autosomal dominant inheritance, but the penetrance of the mutant gene varies widely. There are neuropathic, cardiopathic and ophthalmoleptomeningeal forms of the disease. Most often, the neuropathic form develops, which is characterized by progressive damage to the peripheral and autonomic nervous system. The predominant damage to one or another organ is largely due to the nature of the mutation. Thus, when isoleucine at position 122 is replaced by valine, senile amyloid heart disease develops without neurological manifestations; this mutation is found predominantly in African Americans [11]. Mutations that cause serious heart damage include the substitution of methionine for valine at position 30 (Portuguese variant of familial amyloidosis), the substitution of serine for isoleucine at position 84, and the substitution of alanine for threonine at position 60. Amyloid cardiopathy in hereditary ATTR amyloidosis is less severe, than in AL amyloidosis, but still leads to severe heart failure. In one of the most common types of amyloidosis, secondary AA amyloidosis, clinically significant cardiac damage is rarely detected - in less than 10% of cases [13]. Although AA amyloidosis is systemic in nature, and upon morphological examination amyloid deposits are found in various tissues, in this form of the disease clinical significance is mainly in the kidneys, and to a lesser extent in the liver, spleen and gastrointestinal tract.
Treatment and prevention
In most cases, amyloidosis is treated at home. If there are complications, the patient may be indicated for hospitalization.
Treatment for amyloidosis includes taking medications and following a number of doctor’s recommendations. But in severe cases, the spleen is removed and kidney or liver transplantation may be required.
The list of medications depends on the location of the deposits, the degree of damage to the body, and existing complications. Thus, with secondary amyloidosis, specific treatment of the primary disease is necessary. In addition, medications are prescribed to eliminate symptoms.
Also, the patient is often prescribed a special diet (restriction of protein and salt intake).
There is no specific preventive program for amyloidosis, since the exact causes of the disease are unknown.
Amyloidosis is a complex disease that requires ongoing treatment. A patient with amyloidosis should be regularly monitored by a specialist and undergo examinations to monitor his health. Medical has modern diagnostic equipment, which allows you to make an accurate diagnosis in the shortest possible time. And the center’s experienced, highly qualified specialists will prescribe effective treatment and ensure proper care for the patient.
Cardiac amyloidosis (review of current data)
Nomenclature
Correct nomenclature uses the letter "A" for amyloid, followed by the letter(s) indicating the major deposited protein. For example, light chain amyloidosis is "AL" ("A" for amyloid and "L" for light chain). Transthyretin amyloidosis - "ATTR" ("A" for amyloid and "TTR" for transthyretin). Terms such as "primary amyloidosis", "secondary amyloidosis", "senile amyloidosis" and "familial amyloid cardiomyopathy" are often confusing and should generally be avoided.
The vast majority of cardiac amyloidosis is caused by one of two proteins: light chains or transthyretin. A discussion of presentations and specific treatments for these two subtypes follows.
Light-Chain (AL) Amyloidosis
The most commonly diagnosed form of systemic amyloidosis is AL amyloidosis. A full discussion of the pathophysiology of the disease is beyond the scope of this review, but a brief working understanding is useful to better understand treatment options.
Plasma cells are found primarily in the bone marrow and produce large amounts of antibodies. Antibodies are composed of heavy chains and light chains. When a plasma cell becomes clonal (essentially cancerous), it and its clones typically produce a clonal antibody and a clonal light chain excess associated with that antibody. At this point, there are three possible outcomes:
The plasma cell clone takes over a small portion of the bone marrow and the light chain is excreted harmlessly in the urine. This condition is called monoclonal gammopathy of undetermined significance.
The plasma cell clone occupies a large portion of the bone marrow, which can lead to hypercalcemia, anemia, lytic lesions, and/or renal dysfunction. This condition is called myeloma.
The plasma cell clone produces a light chain that is prone to misfolding into beta-pleated sheets. These light chains circulate in the bloodstream and are deposited in one or more tissues. This condition is called AL amyloidosis.
AL amyloid deposits can occur in virtually any organ, and the pattern of organ involvement varies from patient to patient. Common sites of involvement and associated manifestations include kidney (albuminuria and potential renal failure), liver (elevated alkaline phosphatase and potential liver failure), gastrointestinal (GI) tract (dysphagia, constipation, malabsorption and GI bleeding), tongue (macroglossia) and nerves (peripheral neuropathy and autonomic dysfunction). Patients rarely have clinical involvement of all these organ systems.
Cardiac manifestations include heart failure (HF) (diastolic > systolic) and arrhythmias (tachyarrhythmia/bradyarrhythmia). The key to the diagnosis is ventricular “hypertrophy” observed on echocardiography with inappropriately subtle electrical changes on the electrocardiogram (ECG). B-type natriuretic peptide and N-terminal pro-B-type natriuretic peptide are typically elevated in cardiac amyloidosis, and troponin assays are often chronically positive at low levels (0.1–1 ng/mL) due to ongoing gradual destruction of cardiomyocytes.
Transthyretin (ATTR) amyloidosis
Transthyretin (prealbumin) is an abundant protein produced by the liver and functions as a transporter for thyroxine and retinol. It usually circulates predominantly as a homotetramer, with a small amount of transthyretin circulating in monomeric form. The monomeric form of transthyretin is prone to misfolding and is gradually deposited as amyloid deposits.
There are two main subtypes of ATTR amyloidosis: natural-type ATTR and mutant ATTR. In the case of natural type ATTR, the transthyretin protein is a normal (not mutated) protein and is gradually deposited as amyloid deposits over decades. Although small amounts of deposits may occur in the soft tissue (causing carpal tunnel syndrome) and vasculature, the primary pathological deposits occur in the heart.
Patients with mutant ATTR were born with a pathological mutation in the transthyretin gene, leading to accelerated amyloid deposition. Mutant ATTR is most commonly deposited in the heart and/or nerves, with the pattern of deposition largely dependent on the mutation. In the United States, the most common mutation is the V122I mutation; it is present in 3-3.5% of people of African descent. The exact penetrance of the mutation is unknown and is likely <10% 1 , but it is almost certainly a grossly underdiagnosed form of heart failure in the African American population.
Diagnostics
A definitive diagnosis of amyloidosis requires a biopsy. The subcutaneous tissue of the abdomen is sometimes chosen as a location due to the ease and low risk of injury of the procedure. However, this method suffers from relatively low sensitivity; even in positive cases there are often inadequate amyloid deposits to definitively type the disease (ATTR, AL, etc.). Thus, the general practice at the Stanford Amyloid Center is to biopsy the clinically involved organ (i.e., the heart when cardiac amyloidosis is suspected) because this practice has nearly 100% sensitivity/specificity and almost always allows for a definitive subtype. Subtyping can be done by immunofluorescence or sent to a referral laboratory for mass spectrometry. If there is any doubt about the diagnosis based on immunofluorescence staining, mass spectrometry should be performed.
Laboratory tests for AL amyloidosis include testing for other organ dysfunction (eg, proteinuria, alkaline phosphatase) and direct measurement of circulating light chains. Free light chain analysis may be useful both for assessing the likelihood of diagnosis before biopsy (eg, normal circulating light chains make the diagnosis of AL amyloidosis unlikely) and for monitoring the disease's response to chemotherapy.
For ATTR amyloidosis, it is important to offer genetic testing of the transthyretin gene. The presence of a pathological mutation may influence clinical trial options, predict sites of organ involvement, and have implications for family members.
In any form of cardiac amyloidosis, the dominant imaging feature is the appearance of cardiac “hypertrophy.” The "hypertrophy" on imaging represents amyloid fibril deposition rather than myocyte hypertrophy/hyperplasia, which explains why ECG changes are not apparent. It is important to note that the extent of hypertrophy is variable, so in a patient without classic imaging findings, it is important to maintain clinical suspicion in appropriate circumstances (eg, unexplained cardiac failure in a patient with myeloma or unexplained cardiac failure in a patient with proteinuria/macroglossia).
Cardiac amyloidosis causes abnormal patterns of late gadolinium enhancement on cardiac magnetic resonance (CMR) in both a global transmural and subendocardial distribution. Rise during spin-lattice relaxation of the myocardium on CMR is also common. Transthoracic echocardiography usually reveals basal > predominant apical abnormalities when deformed. Although CMR grade and strain abnormalities have been correlated with patient outcomes, it is currently difficult to say with certainty that they add additional information compared to other prognostic models. In addition, these models are still not specific enough for the diagnosis of amyloidosis to avoid the need for a definitive biopsy-confirmed diagnosis.
The role of repeated assessments of late gadolinium enhancement or spin-lattice relaxation times on CMR or strain assessment using echocardiography is unclear. For AL amyloidosis, it is unclear that this monitoring significantly improves assessment of disease response using changes in serum light chains and cardiac biomarkers.
Forecast
The prognosis for amyloidosis depends primarily on the degree of cardiac damage and, in AL amyloidosis, on the level of circulating light chains. As chemotherapy options have expanded significantly in recent years, the prognosis for AL amyloidosis has also improved markedly, with life expectancy for most patients, including many with significant cardiac damage, measured in years rather than months. The prognosis for ATTR amyloidosis is generally better than for AL amyloidosis, although both forms of the disease still have a high annual mortality rate.
Various staging systems have been proposed for AL amyloidosis, most of which focus primarily on the extent of cardiac involvement. A widely used staging system published in 2004 relied exclusively on two cardiac biomarkers: troponin (T or I) and N-terminal pro-B-type natriuretic peptide. The grade was graded as 1, 2, or 3 depending on whether 0, 1, or 2 biomarkers were elevated. The staging system successfully identified clear survival curves, although overall survival remained disappointing in all 3 groups with median survival of 26.4, 10.5 and 3.5 months in stages 1, 2 and 3, respectively. A revised staging system was published by the same group in 2012, adding the absolute difference in free light chains as a third variable, thus dividing patients into 4 stages. In addition to improving step accuracy, this system has shown improvements overall. Survival across all stages with median survival of 94.1.40.3.14 and 5.8 months for stages 1, 2, 3 and 4 respectively. With continued advances in chemotherapy, it is likely that survival for these patients will continue to improve significantly.
Treatment
Treatment of AL amyloidosis follows two parallel pathways: treating the consequences of organ dysfunction while attempting to slow disease progression by eliminating clonal plasma cells (and thus reducing circulating pathological light chains) through chemotherapy.
Cardiac-specific treatment of all forms of amyloidosis largely involves volume management (diuretics/salt restriction) and arrhythmia management. Neurohormonal antagonists commonly used for HF are often poorly tolerated and counterproductive. In particular, beta blockers, angiotensin converting enzyme inhibitors, and angiotensin receptor blockers often lead to hypotension (due to autonomic dysfunction and the presence of a small left ventricular cavity with an inability to increase stroke volume in response to vasodilation), and beta blockers often worsen bradyarrhythmias . Digoxin binds to amyloid fibrils, resulting in potential digoxin toxicity even at normal blood levels, and should generally be avoided.
Pacemakers are often needed due to the high prevalence of conduction-related diseases (especially ATTR amyloidosis). Atrial fibrillation is common and often poorly tolerated, often requiring cardioversion and/or antiarrhythmic therapy (most commonly amiodarone). Ventricular arrhythmias and sudden cardiac death are also common. Historically, implantable cardioverter defibrillators (ICDs) have been discouraged due to the poor prognosis associated with cardiac amyloidosis, but more recent evidence suggests that ICDs may be an effective part of the treatment strategy in patients with a reasonable prognosis for cardiac amyloidosis ( > 1 year). )
Because cardiac amyloidosis is irreversible and can often be associated with severe symptoms and high mortality rates, cardiovascular transplantation should be considered in well-selected patients. Heart transplantation should be accompanied by a chemotherapy-based strategy to control light chain production. In ATTR amyloidosis, it is important to exclude significant neurological involvement in patients with familial forms. In all cases, we recommend that transplantation be performed only in centers with experience in transplanting patients with amyloidosis.
Chemotherapy options for AL amyloidosis have improved markedly over the past decade, such that most patients can achieve significant reductions (and sometimes complete normalization) of their circulating pathological light chains. Chemotherapy usually involves a combination of several classes of antineoplastic agents, including alkylators (eg, melphalan or cyclophosphamide), steroids (eg, dexamethasone), protease inhibitors (eg, bortezomib or carfilzomib), and/or immunomodulators (eg, lenalidomide). An alternative strategy involves autologous stem cell transplantation using high-dose alkylating agent chemotherapy. Although autologous stem cell transplantation can be an effective treatment for light chain reduction and is used in many centers, the only randomized trial comparing it with standard chemotherapy has shown that autologous stem cell transplantation has inferior outcomes. Standard chemotherapy options for the last decade, our center's practice is to perform autologous stem cell transplantation only in rare cases.
Because ATTR amyloidosis is not a cancer, chemotherapy does not play any role. Although there are currently no approved disease-modifying drugs for the treatment of ATTR amyloidosis, several drugs have been studied and/or are in late-stage trials. These include the following:
Diflunisal. This nonsteroidal anti-inflammatory drug, approved for the treatment of arthritis pain, stabilizes the tetrameric form of transthyretin. A randomized trial demonstrated a slower progression of disease among patients with polyneuropathy due to ATTR mutant amyloidosis. Since nonsteroidal anti-inflammatory drugs are relatively contraindicated in HF, this is unlikely to be a good option for ATTR cardiomyopathy.
Tafamidis. This agent is approved in some parts of the world (Europe and Japan) for ATTR mutant amyloidosis causing polyneuropathy, but is not approved in the United States. A phase 3 study examining its utility in ATTR cardiomyopathy (both natural-type ATTR and mutant ATTR) has completed enrollment and results are pending.
RNA interference. Two agents that act through small interfering RNA or RNA interference (reducing transthyretin production by the liver) are in phase 3 trials for ATTR amyloidosis, including both polyneuropathy and cardiomyopathic forms.
Summary
The paradigm of cardiac amyloidosis has changed markedly over the past decade, with increasing numbers of patients being diagnosed with amyloidosis and notable improvements in therapeutic options. Diagnostic features include unexplained ventricular hypertrophy with subtle ECG changes and unexplained HF occurring with characteristic dysfunction of other organs. Definitive diagnosis requires biopsy and is critical for definitive subtyping of amyloid deposits (eg, AL and ATTR).
For AL amyloidosis, treatment should begin immediately with chemotherapy (our preferred option) or autologous stem cell transplantation. In the case of ATTR amyloidosis, patients should be referred for evaluation for inclusion in one of the ongoing clinical trials. Cardiac management primarily involves diuretics/rhythm control. For selected patients, ICD implantation or heart transplantation should be considered.
Because the disease typically requires multidisciplinary expertise, we recommend referral to an amyloid center for consultation whenever possible.
Treatment
Photo: sodrugestvo.info
The goal of treating amyloidosis, regardless of the type and characteristics of the course of the disease, is to reduce the concentration of special precursor proteins, which subsequently turn into amyloid and are deposited in tissues. In addition, the most important task is to stabilize or restore the functions of internal organs, prevent the development of organ failure and increase the life expectancy of patients.
Symptomatic measures for amyloidosis are aimed at eliminating existing disorders: edema, arrhythmia, hypotension, heart failure, etc. Treatment of amyloidosis is mainly conservative. With the development of complications or a critical decrease in the functions of vital organs, surgical interventions are performed.
Treatment regimens vary depending on the type of disease:
- AA amyloidosis . Anti-inflammatory therapy is recommended in combination with surgical techniques for the development of local purulent processes.
- AL amyloidosis . It is necessary to suppress the clone of plasma cells that secrete special subunits of immunoglobulins (light chains). Stopping the synthesis of such compounds and their accumulation in tissues helps remove excess amyloid and stop the progression of the pathology.
- ATTR amyloidosis . For a long time, the only effective treatment for this type of disease was considered to be a liver transplant that secretes unchanged transthyretin. In recent years, in the early stages of the disease, therapy with a drug from the group of selective transthyretin stabilizers has been used with sufficient success.
Drug therapy
In the secondary form of the disease, pathogenetic therapy is required, aimed at eliminating or alleviating the course of the underlying disease. The list of medications, doses and duration of courses are determined individually, taking into account the nature and severity of the causative pathology. AA amyloidosis requires long-term, sometimes lifelong, basic therapy, which is supplemented with antibacterial drugs when complications develop.
High-dose chemotherapy is indicated for patients with AL amyloidosis. Achievement of remission is assessed based on laboratory parameters. Taking into account the existing symptoms in any form of amyloidosis, medications are used to eliminate disturbances in the functioning of the heart and blood vessels, correct blood pressure and intestinal motility, and combat pain.
Surgery
In case of life-threatening cardiac arrhythmias, a pacemaker is required. With the development of local purulent processes against the background of AA amyloidosis, opening and drainage of abscesses in soft tissues, lungs, and joints is indicated. For ATTR amyloidosis, liver transplantation is performed. The technique is especially effective in people under 50 years of age with no complications from the cardiovascular system.
Since systemic amyloidosis often affects the kidneys with the development of renal failure, a number of patients require transplantation of this organ. Surgery is recommended in the absence of gross changes in the heart and gastrointestinal tract. The five-year survival rate after transplantation is about 65%, which is comparable to the rates for patients with chronic renal failure due to other diseases. A third of recipients of a transplanted kidney also subsequently develop amyloidosis, but transplant rejection as a result of amyloid deposition occurs in only 2-3% of cases.
Renal replacement therapy
Chronic renal failure is one of the leading causes of death in patients with systemic amyloidosis. Taking into account this circumstance, in case of serious impairment of kidney function, methods of extracorporeal blood purification become highly important. According to experts, the preferred option is peritoneal dialysis, which carries a lower risk of pressure drop during the procedure. Life expectancy for hemodialysis and peritoneal dialysis is the same as for people with other kidney diseases. Good or satisfactory clinical and laboratory results of renal replacement therapy are observed in 60% of cases.
Morphology and pathogenesis
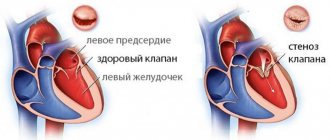
Amyloid deposition leads to significant concentric thickening of the myocardium, and dilatation of the ventricular cavities is not typical for AS. Amyloid infiltration of the myocardium is accompanied by a deterioration in its mechanical properties and leads to a significant decrease in contractility in combination with severe restrictive disorders of diastolic function. Modern research methods have shown that thickening of the myocardial walls in AS does not always correlate with impaired systolic and diastolic functions. Conventionally, three subtypes of AS can be distinguished: in patients with AL, hereditary ATTR and acquired senile ATTR amyloidosis [16].
In all cases, amyloid is found in the myocardial interstitium in the form of diffuse or nodular deposits. The most common findings are diffuse thickening of the interventricular septum (> 80% of patients) and the posterior wall of the LV. Isolated thickening of the wall of the right ventricle (without thickening of the LV wall) is rare - in 6% of cases, and thickening of the walls of both ventricles - in 40-80% of cases [3, 18]. Thickening of the interatrial septum is observed in 40% of patients. The change in the shape of the LV occurs according to the type of concentric hypertrophy; ventricular dilatation does not develop [19]. The combination of thickening of the myocardial walls in more than half of the patients with low amplitude of the ventricular complex on the electrocardiogram (ECG) in the chest leads (less than 10 mm) and limb leads (less than 5 mm) allows us to confirm the pseudohypertrophic nature of myocardial changes. According to C. Rapezzi et al., the thickness of the myocardial walls in patients with ATTR amyloidosis (especially senile) was significantly greater than in patients with AL amyloidosis [16, 20]. At the same time, patients with AL amyloidosis showed a more significant decrease in the amplitude of the ventricular complex and, accordingly, a lower ratio of QRS amplitude to LV wall thickness. This may indicate that the decrease in QRS amplitude in patients with AL amyloidosis is more related to damage to cardiomyocytes than to interstitial amyloid deposition. It is known that in the pathogenesis of organ damage in patients with AL amyloidosis, the direct toxic effect of free light chains of immunoglobulins on cells is of great importance [16, 18]. In patients with different types of AS, significant vacuolization of cardiomyocytes is detected. Studies in recent years have shown [24] that it is the vacuolization of cardiomyocytes, and not amyloid deposits, that is responsible for the increased echogenicity of the granular type, characteristic of AS and first described by A. Siqueira-Filho et al. [19]. Thus, this ultrasound property of amyloid heart is not specific and cannot be used as a diagnostic criterion.
Damage to cardiomyocytes in patients with AS may be reflected in increased concentrations of troponins and natriuretic peptides. The expression of atrial and medullary nitriuretic peptides has been shown to be increased in ventricular cardiomyocytes from patients with AS, particularly in areas adjacent to amyloid deposits [25]. Although these substances are not specific markers of amyloid cardiac damage, they are considered highly sensitive indicators and, therefore, an important prognostic factor in systemic amyloidosis [26].
Amyloid can be deposited in the area of the heart valves, often causing their thickening, noticeable on an echocardiogram [18, 27]. Thickening of the papillary muscles has been described [28], but valve function is often preserved.
Amyloid can damage the conduction system of the heart; cases of amyloid masses penetrating directly into the sinoatrial node have been described. Conduction disorders are most often represented by incomplete blockade of the anterior branch of the left bundle branch (20%), complete blockade of the right (4–19%) or left bundle branch (2–7%), and first degree atrioventricular block (18–33%) [16, 18]. Conduction disturbances, as a rule, occur in senile AS, which may be due to the longer course of this variant of the disease [3]. Amyloid deposition in the area of adrenergic synapses disrupts the neurohumoral regulation of heart function [29], which may be one of the risk factors for tachyarrhythmias. In 5–27% of patients, atrial fibrillation is detected; more rare rhythm disturbances are ventricular tachycardia and junctional rhythm. Some changes in the ECG occur in almost all (> 90%) patients with AS.
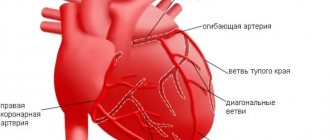
A distinctive property of AL amyloidosis is its deposition in the walls of the coronary arteries [15], which can cause myocardial ischemia [30]. Some patients complain of angina pectoris [31], although changes in the angiogram may be absent [32]. Myocardial infarction may develop. Pathological Q-waves and changes in repolarization without clinical signs of myocardial infarction or indication of it in the anamnesis are found in more than half of the patients, which allows these changes to be interpreted as pseudo-infarction. It appears that mimicking infarct changes is associated with nodular deposition of electrically inactive amyloid, which creates a scar effect. In the later stages of the disease, pericardial effusion is detected in almost half of patients [18].
Quantitative assessment of the total mass of amyloid deposits in the heart is possible using SAP scintigraphy. SAP is a normal plasma glycoprotein whose levels increase dramatically in amyloid deposits of any type as a result of reversible calcium-dependent binding to the amyloid fibril. After intravenous administration, the radiotracer labeled with SAP is distributed between the circulating and amyloid-bound SAP pools in proportion to their volume. In this way, images can be obtained for qualitative and quantitative assessment of amyloid deposits. The method is especially useful in monitoring the course of amyloidosis, including for the purpose of assessing the effectiveness of treatment.
Course of the disease and prognosis
Experts say that the main cause of death in patients is chronic renal failure, which occurs as a result of reactive amyloidosis, which is characterized as an inflammatory infectious process. There is also information that patients with an outbreak of reactive amyloidosis die as a result of an infectious intestinal disease and diarrhea.
Statistics indicate that even with timely detection and treatment of the disease, the average survival rate of patients with renal failure is no more than one and a half years from the date of examination.
Today, leading specialists prolong the lives of patients through bone marrow transplantation, and also perform surgical procedures for kidney transplantation. #diagnosis #urology kidney problems
Classification of renal amyloidosis
Systematization of the disease is based on parameters such as etiology and pathogenesis. Based on them, five of its forms are distinguished.
| Form | Characteristics |
| Congenital | AL amyloidosis, the development of which was provoked by the appearance and accumulation of light chains of immunoglobulins in the body tissues. The etiology of the disease still remains unclear, as well as the mechanism of its development. |
| Acquired | AA amyloidosis is secondary, caused by a number of chronic inflammatory processes in which the liver begins to intensively produce the acute phase protein alpha globulin. |
| Family | AF amyloidosis (or "Mediterranean fever") is hereditary. Residents of the Mediterranean (Greeks, Arabs, Armenians) suffer from it. The disease is based on a genetic defect that causes a disruption in the production of fibrillar proteins. |
| Senile | ASC1 amyloidosis develops in elderly patients due to impaired metabolism of serum prealbumin. According to medical statistics, the disease affects about 80% of patients over 80 years of age. |
| Local tumor-like | AE amyloidosis can be triggered by neoplasms of the endocrine system, type 2 diabetes mellitus, and senile dementia of the Alzheimer's type. |
Diagnostics
Photo: pluska.sk
Modern methods for diagnosing amyloidosis
Amyloidosis is a disease that manifests itself as a pathology of protein metabolism and the performance of the immune system. Amyloid (protein-saccharide complex), which appears as a result of this disorder, can be deposited in the cells of any tissue of the body. As the disease progresses, it gradually replaces healthy cells, and the organ ceases to function. In severe forms of the pathology, multiple organ failure occurs (damage to 50% or more organs), leading to death.
The hereditary form of the disease is observed in the Mediterranean area, as well as in people of Jewish and Armenian nationality. Among men, this disease occurs twice as often.
The most common forms of the disease include nephropathic, in which deposits are observed in the kidneys, and systemic, in which amyloid deposits are found in many organs.
Amyloid deposited in different organs differs in structure. There are about 15 types in total, differing in structure and composition. They are based on two types:
- AA amyloid. Chronic inflammatory diseases cause high plasma levels of SAA protein and maintain them for a long period. When the protein is incompletely broken down, fibrillar AA amyloid is formed.
- AL amyloid. Proteins of this type appear during the cleavage of amyloidoblasts (degenerated plasma cells). They are abnormal immunoglobulin compounds.
- The appearance of other types of amyloid is determined by the form of amyloidosis.
Types of disease
Amyloidosis occurs both independently and as a concomitant disease. Diagnosis of amyloidosis distinguishes several types:
Primary or idiopathic. With this type of deposits, deposits are found in all internal organs. It is impossible to determine the exact cause of the appearance. Multiple abnormalities are observed in the cells of the immune system with the accumulation of AL-amyloid in the skin, tissues of the muscular, nervous and cardiovascular systems. Its cause may be plasmacytoma (myeloma), a malignant tumor pathology.
Symptoms of primary amyloidosis:
- Myasthenia gravis with subsequent muscle atrophy;
- Dyspepsia and diarrhea;
- Pathology of the genitourinary and reproductive system;
- Damage to the organs of vision.
Secondary
It is a complication of any inflammatory disease. The reason for its appearance may be:
- Chronic infectious diseases: pyelonephritis, tuberculosis, bronchiectasis, malaria, syphilis or leprosy (leprosy);
- Chronic purulent diseases: osteomyelitis, purulent ulcers and wounds;
- Ulcerative colitis – inflammation of the colon;
- Tumor lesions of the hematopoietic organs: leukemia, lymphogranulomatosis, etc.;
- Rheumatological pathology: various arthritis, etc.
Secondary amyloidosis forms in the internal organs. There is a disruption in the functioning of organs with the greatest deposition of amyloid - in the area of the kidneys, spleen, liver or lymph nodes. Subsequently, the damage spreads to other organs, followed by death.
Hereditary
This form is formed due to genetic abnormalities in the cells of the immune system, leading to the appearance of amyloidoblasts. A similar pathology is diagnosed in some national groups or in a certain geographical area. Hereditary forms include:
- Periodic disease - familial Mediterranean fever;
- Familial nephropathic or English amyloidosis;
- Hereditary neuropathic amyloidosis - Portuguese, American or Finnish;
- Hereditary cardiopathic or Danish amyloidosis.
Senile
A systematic approach allows us to identify this pathology in people over 80 years of age. It includes:
- Cerebral or cerebral amyloidosis. Diagnosed with Alzheimer's disease;
- Amyloidosis of the heart. Affects the heart muscle. Deposits also form in the lungs, liver and pancreas.
Tumor
In this case, amyloidosis develops locally in organs with a pronounced malignant process. It is caused by medullary thyroid cancer or pancreatic islet tumor.
Hemodialysis
During hemodialysis prescribed to patients with renal failure, the level of B 2 -microglobulin in the blood gradually increases. This protein, when interacting with nucleoproteins, settles in the kidney tissues.
Diagnosis of amyloidosis
To diagnose amyloidosis, the patient is prescribed a number of different tests. This includes a general blood and urine test, blood biochemistry, ultrasound of internal organs, biopsy and genetic research.
General blood analysis
Determines abnormalities specific to amyloidosis. In the final stages of the disease, this study helps to identify the damaged organ.
General urine analysis
Diagnosis of renal amyloidosis shows the likelihood of developing inflammatory processes in the kidneys.
In case of kidney pathology, the following is detected:
- Proteinuria – protein content in urine over 3 g/l;
- Hematuria – detection of red blood cells in the urine;
- Leukocyturia – the presence of leukocytes in the urine;
- Cylindruria - the content in the urine of casts formed during amyloidosis from proteins, kidney epithelial cells, leukocytes and erythrocytes;
- Decreased urine density.
Blood chemistry
It makes it possible to assess the general condition of the body and determine the cause of amyloidosis. This analysis determines:
- Proteins of the general phase of inflammation, produced by the liver or certain leukocytes during the inflammatory process. Particular attention should be paid to the amount of fibrinogen.
- Liver tests indicate the condition of this organ.
- Increased cholesterol levels are a sign of nephrotic syndrome.
- A decrease in protein levels indicates nephrotic syndrome or liver failure.
- An increase in urea and creatinine levels is an indicator of renal dysfunction in amyloidosis.
Ultrasonography
This method makes it possible to determine the structure and structure of the tissues of internal organs, the degree and distribution of pathological processes.
Diagnosis of amyloidosis using ultrasound shows:
- Hardening and change in the size of the kidneys;
- Presence of cysts in the kidneys;
- Compaction and enlargement of the spleen and liver, accompanied by pathology of blood flow;
- Hypertrophy of the heart muscle;
- The presence of amyloid deposits in the walls of the main blood vessels;
- An increase in the volume of fluid in various cavities of the body - ascites, hydropericardium or hydrothorax.
Biopsy
Removal of a small part of tissue for examination using special methods. Its use makes it possible to diagnose amyloidosis in 90% of cases. Tissue from muscles, internal organs, and mucous membranes is taken for examination.
Genetic research
It is carried out if there is a possibility of developing hereditary amyloidosis. The patient's genetic material is taken for research and checked for the presence of genetic abnormalities in some chromosomes. If a pathology is detected, it is recommended that all blood relatives of the patient undergo examination to identify this disease in them.
Classification of kidney disease
There are primary and secondary renal amyloidosis. Primary amyloidosis is characterized as a disease that occurs as a result of improper adhesion of immunoglobulins, which settle not only in organs, but also on the walls of blood vessels, and also affect the composition of the blood. Moreover, primary amyloidosis is difficult to diagnose because it has a large number of common symptoms with other serious diseases.
Secondary amyloidosis develops as a result of chronic infections and diseases such as acquired osteomyelitis, spondyloarthritis, colitis, cancer, sclerosis, rheumatoid arthritis and tuberculosis. Secondary amyloidosis can develop in old age, as well as as a genetically determined factor.
All forms of the disease are characterized by:
- arise and continue to develop mainly during early childhood;
- occur with periodic exacerbation;
- the presence of pain attacks;
- unsuccessful treatment;
- damage only to the kidneys.
Four stages of the disease:
- The latent stage of the disease is characterized by asymptomatic progression. Only swelling and signs of sclerosis are observed. The stage can last approximately 6 years. Over the past 2 years, there is a possibility that the first signs of the underlying disease (tuberculosis, arthritis, malignant tumor) will appear.
- The albuminuric stage of the disease is caused by the appearance of amyloid on the walls of blood vessels. At this stage, the patient develops sclerosis, lymphostasis, and the kidneys increase in size. The stage lasts approximately 12 years.
- Edema stage. At this stage, amyloids completely affect the nephron. The main internal organs are enlarged, deep sclerosis, severe swelling and arterial hypertension are noted, and signs of focal damage to the intestinal tract are observed. The stage lasts no more than 7 years.
- Azotemic stage - a wrinkled, deformed kidney is observed, having a high density and scars. The patient has severe renal failure and suffers from low blood pressure.
Clinical picture and prognosis
Amyloid damage to the heart in the early stages can be asymptomatic, manifested only by thickening of the LV wall on echocardiography [37]. Subsequently, clinical manifestations of HF with blood stagnation develop mainly in the systemic circulation [18]. Progressive shortness of breath and decreased exercise tolerance are observed in more than half of the patients and are accompanied by signs of pulmonary hypertension [2, 38]. In AL and senile ATTR amyloidosis, HF is more common (NYHA functional classes III–IV in more than half of the patients) than in hereditary ATTR amyloidosis [3, 16]. The systemic nature of the lesion is characteristic of AL amyloidosis. The majority develop damage to the kidneys (nephrotic syndrome, renal failure), liver (giant hepatomegaly, intrahepatic cholestasis), spleen (splenomegaly), muscles (macroglossia, muscle pseudohypertrophy), skin (periorbital purpura, amyloid plaques), peripheral and autonomic nervous system. Rarely, thromboembolism of the coronary arteries with amyloid masses develops, leading to death [39]. Thrombosis of various vessels has also been described [18].
The diagnosis of systemic amyloidosis is made by histological examination of tissue obtained from a biopsy. More often the wall of the rectum, stomach, oral cavity, subcutaneous fatty tissue, kidney and liver tissue are examined. Determination of the variant of amyloidosis is carried out using the immunohistochemical method; in case of hereditary amyloidosis, DNA analysis is done. If amyloidosis is limited to the heart, such as in hereditary ATTR amyloidosis with isoleucine mutation at position 122, then the only diagnostic method is myocardial biopsy. However, in most other cases, the diagnosis of AS can be established when amyloid is detected in a biopsy of another organ or tissue and the patient has a thickening of the LV wall of more than 12 mm according to echocardiography [40].
The most severe prognosis is for AL amyloidosis, and somewhat more favorable for ATTR amyloidosis. Moreover, the prognosis is determined precisely by heart damage. In a study of 232 patients with AL amyloidosis with cardiac involvement, their average life expectancy after diagnosis was 1 year. The presence of HF reduced survival: 0.75 versus 2.34 years in patients without HF.
Traditionally, cardiac predictors of poor prognosis, in addition to the severity of HF and restrictive disorders of LV diastolic function, are considered to be thickening and increase in LV mass with a decrease in ejection fraction [18, 41]. At the same time, C. Rapezzi et al. revealed the most significant thickening of the myocardial wall and increase in myocardial mass in patients with senile ATTR amyloidosis, in which relatively slow progression of AS is observed [16]. It is noteworthy that moderate thickening of the myocardial walls in patients with AL amyloidosis who participated in this study was combined with a significant decrease in the voltage of the ventricular ECG complexes. The authors associated these ECG changes with the direct toxic effect of immunoglobulin light chains on cardiomyocytes. At the same time, massive deposits of amyloid in patients with senile ATTR amyloidosis apparently do not have such an adverse effect on myocardial cells and, therefore, have less impact on the survival of patients. In another study of 57 patients with AL and ATTR amyloidosis, low ECG voltage was also associated with a worse prognosis [42].
Currently, high blood levels of markers of cardiomyocyte damage - troponins T and I, and N-terminal protein-brain natriuretic factor (NT-proBNP) at the time of diagnosis of AS are considered an important predictor of poor prognosis. Thus, in 261 patients with a newly diagnosed diagnosis, the average life expectancy with an increase in the level of troponins I or T was 6 and 8 months, respectively, and in the absence of troponins I or T in the blood - 21 and 22 months [43]. In this study, the prognostic value of serum troponin levels was higher than that of congestive heart failure and echocardiographic parameters. The possibility of analyzing the dynamics of NT-proBNP levels after treatment to assess the prognosis of AS is being studied. In AL amyloidosis, a 30% reduction in NT-proBNP levels after chemotherapy was associated with improved prognosis [44].
An important prognostic factor is also the presence and severity of extracardiac manifestations of systemic amyloidosis (orthostatic hypotension, complications of nephrotic syndrome and motor diarrhea, renal failure, etc.).
Thus, among the variants of AS, AL amyloidosis has the most severe and rapidly progressive course, in which the development of HF depends little on the degree of thickening of the myocardial walls and is not always combined with restrictive disorders of diastolic function. The progression of AS in these patients is largely due to damage to the cardiomyocyte. In patients with senile ATTR amyloidosis, a relatively mild course of cardiopathy is observed, despite significant structural (severe pseudohypertrophy) and functional (restrictive hemodynamic disorders) disorders. An intermediate position is occupied by hereditary ATTR amyloidosis, in which the symptoms of cardiopathy are close to those of AL amyloidosis of the heart, but are less malignant.
Cardiac dysfunction
Intracardiac hemodynamic disturbances in AS are caused mainly by diastolic dysfunction; the decrease in myocardial contractility is usually insignificant. In a study that included 223 patients with AS, the mean LV ejection fraction was 52.5 ± 13.1% in patients with AL amyloidosis and 58 ± 13% in patients with hereditary ATTR amyloidosis. It was significantly lower only among patients with senile ATTR amyloidosis: 44.2 ± 15.4% [16]. A decrease in ejection fraction of less than 40% was noted in 40% of patients with senile ATTR amyloidosis and only in 22% of patients with AL amyloidosis and 8% of patients with hereditary ATTR amyloidosis. Other researchers have described similar changes in contractile function [18].
Diastolic dysfunction of varying degrees is detected in almost all patients. Early in the course of the disease, amyloid deposits impair isovolumic relaxation of the myocardium, resulting in a decrease in the rate of early diastolic LV filling (E) and an increase in late diastolic flow (A). Thus, a decrease in the E/A ratio (type 1 diastolic dysfunction) is an early sign of heart damage. As AS progresses, the myocardial wall becomes stiffer, the pressure in the left atrium increases, which leads to an increase in the rate of early diastolic filling and, thus, to a pseudo-normalization of the E/A ratio (type 2 diastolic dysfunction) [2]. With a further increase in the stiffness of the heart wall and an increase in end-diastolic pressure, a significant decrease in late diastolic flow occurs, which makes it possible to identify a decrease in myocardial compliance (3rd restrictive type of diastolic dysfunction). Restrictive changes are often accompanied by dilatation of both atria [3, 18].
Although restrictive disorders of diastolic function are considered the most typical for AS, research data show the possibility of developing other disorders of intracardiac hemodynamics [34–36].