- Advantages
- Doctors
- Contacts
- Licenses
Advantages
- The latest, constantly updated equipment
- Interest-free installments for all services
- Online consultations with an ENT doctor
- Visit of an ENT doctor to your home
- Friendly and qualified staff
- 24/7 ENT assistance
Pulmonary hypertension is a cardiovascular disease characterized by high blood pressure in the pulmonary artery. Pressure increases due to narrowing of the walls of the arteries leading from the heart to the lungs. This causes the heart to work harder to pump blood through the lungs. The extra effort eventually leads to weakening of the heart muscle and the development of serious heart failure.
Pulmonary hypertension can be life-threatening. Although some types have no cure, when diagnosed early, treatment for pulmonary hypertension reduces symptoms and improves quality of life.
The code for pulmonary hypertension according to ICD 10 is I27.0.
Introduction
Pulmonary hypertension (PH) is a disease associated with increased pressure in the blood vessels of the lungs.
In most cases it is secondary, but idiopathic forms of PH also occur. Idiopathic (primary) PH is characterized by the absence of any known causes of the disease. It occurs in 1–2 cases per 1 million population. 6% of such patients will have familial PH [1]. And although the incidence of PH in various diseases requires clarification, it is known that, for example, with scleroderma it ranges from 2 to 35%, with portal hypertension - 2-4%, with HIV - 0.1-0.6% [1].
In PH, the pulmonary vessels are in spasm for a long time, their wall is hypertrophied, which ultimately leads to fibrosis and a decrease in the vascular bed. It is this mechanism that underlies the development of right ventricular overload and failure. The main symptoms of PH are shortness of breath, weakness, chest discomfort and fainting. Diagnosis of this disease is quite simple - measuring the average pressure in the pulmonary artery. In 2004, the upper limit of this indicator at rest was defined as 25 mmHg. Art., and with load - 30 mm Hg. Art. [1]. If the average pressure in the pulmonary artery is higher than these values, an examination is necessary to identify the cause of PH and decide on treatment for the disease.
However, in the last decade, the PH paradigm has changed, and in 2018, at the 6th International Symposium on PH, the definition and classification of the disease, as well as approaches to its therapy, were updated [2].
Symptoms of pulmonary hypertension
Signs of pulmonary hypertension appear slowly. They may go unnoticed for months or even years. Symptoms get worse (or more pronounced) as the disease progresses. These include:
- shortness of breath, first during physical activity, and then at rest;
- fatigue;
- dizziness;
- fainting;
- chest pressure or pain;
- swelling of the ankles, legs and eventually the abdomen (ascites);
- bluish color of lips and skin (cyanosis);
- increased pulse and heart rate.
The role of regional centers and LG offices
The main task of regional centers is to determine the PH group (according to the classification), its hemodynamic parameters, and also decide on the prescription of specific therapy.
Patients who apply to these centers should be further examined. PH is most common in the population due to pathology of the left side of the heart and pathology of the lungs. This determines the diagnostic search. All patients should undergo an echocardiogram according to a special protocol, an electrocardiogram (ECG), a chest x-ray, a complete blood count, a biochemical blood test, blood gas determination, NT-proBNP (natriuretic peptide) level, spirometry (if not previously done on an outpatient basis). outpatient department), 6-minute walk test; according to indications - cardiorespiratory exercise test, computed tomography, coronary angiography, etc.
If a patient has a pathology of the left heart or lung disease in the absence of severe right ventricular dysfunction and severe PH, he should be recommended periodic echocardiography monitoring (usually every 6-12 months or if the condition worsens) and treatment of the underlying disease should be started. .
It should be remembered that the patient may have a mixed etiology of PH, therefore, if the severity of the underlying disease and right ventricular dysfunction do not correspond, he should be referred to the Federal Expert Center.
If a patient is suspected of having CTEPH, he needs ventilation-perfusion lung scintigraphy. If such equipment is not available, the possibility of sending it to MSCT angiography (multispiral computed tomography), which is available in most regional centers, should be considered. Also, according to indications, such patients are diagnosed with coagulopathies [1].
If systemic connective tissue diseases are suspected, primarily systemic scleroderma, tests for antinuclear and anticentromere antibodies, as well as antibodies to Scl-70 (topoisomerase I) are necessary [1].
According to the hemodynamic definition, PH can be precapillary, isolated postcapillary and combined pre- and postcapillary (Table 2) [2]. To diagnose it, it is necessary to carry out invasive catheterization of the right heart, which is available in expert or large regional medical centers. In order to evaluate the possibility of prescribing calcium channel blockers to patients during this procedure, a vasoreactive test is also performed [2].
After diagnosing the PH group, all patients need to have their risk of adverse outcomes assessed over the next 12 months. (Table 3). It is recommended to determine the patient's risk at each doctor's visit. It should be noted that this algorithm was developed based on data obtained from studies in patients with idiopathic PAH and has not been validated for other forms of PH [1].
Disease prognosis
The prognosis of pulmonary hypertension is unfavorable. It will not be possible to achieve a complete recovery. If the patient receives treatment, then heart failure leading to death will still occur, but the patient will still be able to prolong the life.
- If the cause of pulmonary hypertension is systemic scleroderma, then the prognosis is extremely unfavorable. During the disease, normal organ tissue degenerates into connective tissue. As a result, a person dies within the first year.
- With idiopathic pulmonary hypertension, the prognosis is slightly improved. Such patients can live on average three years after diagnosis.
- If a heart defect leads to pulmonary hypertension, the patient is referred for surgery. The five-year survival rate of such patients is 40-44%.
- If, against the background of pulmonary hypertension, heart failure rapidly increases with damage to the right ventricle of the heart, then death will occur within 2 years after the manifestation of the disease.
- If pulmonary hypertension has an uncomplicated course and can be corrected with medication, then about 67% of patients cross the 5-year mark.
PH therapy
The treatment of patients with PH is based not only on the prescription of specific therapy, but also on a number of other measures that can improve the patient’s prognosis.
It is advisable to individually select the level of physical activity for patients with PH. It should not cause severe shortness of breath, dizziness, syncope, or chest pain [4]. Vaccination against influenza and pneumococcus should be recommended to all patients with PAH. When traveling by air, due to the high risk of developing vasoconstriction due to hypoxia, it is necessary to provide oxygen for the patient. Women with PAH are recommended to terminate pregnancy due to the high risk of adverse outcomes [5].
Also, patients with PH need maintenance therapy aimed at treating heart failure and improving the prognosis.
For anemia of any degree, correction of hemoglobin levels is necessary, since individuals with PH are highly sensitive to its decrease. In case of erythrocytosis with a hematocrit of more than 65% (in the presence of symptoms - headaches, impaired concentration), developed against the background of long-term hypoxia, phlebotomy is possible. In other cases, phlebotomy should be avoided [1].
Long-term oxygen therapy is indicated when oxygen saturation decreases to less than 90%. It is most effective in patients with PH due to lung disease. In people with Eisenmenger syndrome, according to studies, oxygen therapy does not affect hematological parameters, quality of life and survival [6].
In case of decompensation of right ventricular failure, patients are prescribed diuretic therapy in accordance with the recommendations for the management of patients with chronic heart failure (CHF) [7]. Renal function and blood electrolytes should be regularly assessed in order to promptly adjust the dose of diuretics.
Angiotensin-converting enzyme inhibitors (ACEIs) and beta blockers in patients with PAH have been shown to have little effect and are therefore not currently used. In addition, the use of these drugs, even in small doses, can lead to the development of hypotension and worsening of right ventricular failure [8].
Oral anticoagulants (OAC) can be used in some patients with PH. In idiopathic PAH they significantly improve survival (hazard ratio 0.79, 95% confidence interval 0.66–0.94). They are also indicated in patients with CTEPH, inherited PAH and associated PAH while taking anorectics. In other forms of PH, in particular PAH associated with systemic connective tissue diseases, portopulmonary hypertension and PAH associated with congenital heart defects (CHD), the risk of bleeding may be increased, which requires an individual approach to the prescription of this group of drugs [1].
When myocardial contractility is reduced in patients with PAH, cardiac glycosides may be useful. Intravenous administration of digoxin in this group of patients causes a moderate increase in cardiac output. Patients with atrial fibrillation/flutter can, in some cases, take cardiac glycosides to control heart rate. The use of inotropic drugs (eg, dopamine), as in left ventricular failure, may be effective in patients with PH [9].
If it is necessary to prescribe antiarrhythmic drugs, primarily for atrial fibrillation/flutter, preference should be given to drugs without a negative inotropic effect, such as amiodarone [1].
If a patient has PH associated with damage to the left side of the heart or lung diseases, the patient, as a rule, is not indicated for specific therapy [8].
Development mechanism
The primary form of pulmonary hypertension is characterized by the following processes:
- Smooth muscle hypertrophy (accelerated growth of smooth muscle cell structures).
- Variable vasoconstriction (narrowing or spasm of blood vessels).
- Remodeling of the vascular wall (decreased elasticity, thickening of the vascular lumen, as a result of the formation of blood clots).
Changes in the anatomical structure of blood vessels lead to impaired blood circulation, which causes increased pulmonary pressure.
Important! The progression of the pathological process becomes a provoking factor for right ventricular dysfunction, resulting in right ventricular failure.
Specific therapy for PH
The decision to prescribe specific therapy should be made only after catheterization of the right heart with a test for vasoreactivity. If the test result is positive (about 10% of patients with idiopathic PAH), calcium channel blockers are used in high doses. However, only 50% of patients have a stable hemodynamic response to this group of drugs. Features of the prescription and dose of calcium channel blockers for the treatment of PH are presented in Table 4. It should be remembered that the use of this group of drugs empirically, without performing a vasoreactive test, can worsen the clinical condition and prognosis of the patient [10]. Prescribing standard doses of calcium channel blockers is possible only if there are other indications (for example, Raynaud's syndrome, etc.) [1].
In case of a negative test for vasoreactivity, the patient is shown other groups of drugs:
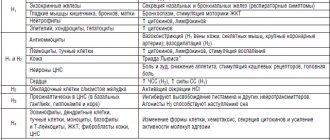
endothelin receptor antagonists (bosentan, ambrisentan, macitentan);
prostanoids (iloprost, epoprostenol (not registered in the Russian Federation));
phosphodiesterase-5 inhibitors (sildenafil, tadalafil, vardenafil);
stimulators of soluble guanylate cyclase (riociguat);
selective prostacyclin IP receptor agonists (selexipag);
combination therapy.
They are expensive and can only be prescribed by an expert center for the treatment of patients with PH. According to the treatment algorithm for PAH in patients with moderate and high risk, the use of combination therapy is preferable (Fig. 1, 2). In this case, in case of insufficient effectiveness, a third drug is added to the combination [10].
The selection of therapy for a patient with PH is carried out by an expert center. After discharge, he needs to report to a regional center or office for patients with PH, where the need for specific therapy will be confirmed by a medical commission. The conclusion of the medical commission and medical documents are then sent to the regional Ministry of Health, where the issue of issuing the necessary medications to the patient is decided.
It should be noted that control of specific therapy is carried out not only by the expert center, but also by regional centers. All complex issues of patient management in the region can be resolved using telemedicine technologies.
Taking into account the high cost of PH therapy, the emergence of high-quality generics can significantly reduce the cost of patient treatment while achieving comparable effectiveness. In 2022, a new drug from the class of endothelin receptor antagonists (bosentan) was registered in the Russian Federation - Bosenex, which has proven bioequivalence with the original drug. However, bioequivalence is not reliable evidence of the effectiveness of a drug.
From December 1, 2017 to September 31, 2018, the Federal State Budgetary Institution National Medical Research Center of Cardiology of the Russian Ministry of Health conducted a study of the safety and effectiveness of Bozenex therapy in patients with PAH for 24 weeks. [eleven]. The study included 42 patients who were initiated on bosentan therapy; 63.6% had not previously received PAH therapy; in 36.4%, the drug was added to therapy with sildenafil at a dose of 60 mg/day. By the 24th week. there was a decrease in the proportion of patients with 3 FC from 55% to 30%, an increase in 2 FC from 45% to 55%, and the appearance of patients with 1 FC (15%); the distance in the 6-minute walk test increased by 52.1 m. During catheterization of the right heart, a positive change in the average pressure in the pulmonary artery (-6.7 mm Hg), average pressure in the right atrium (-1.6 mmHg with achievement of normal values) and pulmonary vascular resistance (-293.2 dyne × s × cm-5) (p <0.05). Bosenex therapy was well tolerated and was not accompanied by clinically significant adverse events [11].
Another proof of the good clinical effectiveness of the drug is a clinical case.
Why is it developing?
Pulmonary arterial hypertension occurs in two types, depending on the cause of its occurrence.
Primary or idiopathic
The etiology of the disease has not yet been precisely established, and the form of the pathology is quite rare. Severe idiopathic hypertension is first diagnosed in newborns, having a hereditary form. Pathology can be transmitted not only in a direct line from the mother or father, but also from grandparents or even great-grandparents.
During diagnosis, children with a similar diagnosis are found to have atherosclerotic changes in the cavity of the pulmonary artery and an increase in the mass of the right cardiac ventricle. If a child or adult who first develops pulmonary hypertension syndrome is not given immediate assistance, he will remain disabled, and complete ignorance of the symptoms can lead to the death of the patient.
Secondary
It develops more often in adults against the background of concomitant diseases. Secondary pulmonary hypertension is observed in pathologies occurring against the background of insufficient functioning of the left ventricle, HIV infection and heart defects of congenital etiology. The risk of developing the disease increases for people with connective tissue pathologies - lupus erythematosus, systemic scleroderma.
With acquired heart defects, after thromboembolism has been transferred to the pulmonary artery cavity, there is also a high risk of signs of increased blood pressure in the vascular bed of the pulmonary arteries. In lung diseases, pathology can also manifest itself, but usually does not reach an advanced stage. Potential dangers are those conditions that increase the risk of developing pulmonary hypertension.
This is taking drugs from certain drug groups (Aminorex, Amphetamines, Cocaine, Fenfluramine, Chemotherapy drugs); oral contraceptives, antidepressants, and estrogen drugs are considered less dangerous in this regard. Diseases that increase the chances of developing pulmonary hypertension are HIV infection, the presence of congenital shunts located between the systemic and pulmonary capillaries, severe diseases of the thyroid gland and liver, genetic mutations and metabolic disorders.
No ads 1
Clinical observation
Patient K., 42 years old, applied for an outpatient appointment at the PH office of a regional center with complaints of shortness of breath, weakness, and periodic chest pain not associated with physical activity. 10 years ago, a congenital heart defect, multiple ventricular septal defects, and a bicuspid aortic valve were discovered. Eisenmenger syndrome. High LH. Moderate aortic valve stenosis. CHF stage 2A, FC 3. Right-sided scoliosis.
Upon examination, the condition is of moderate severity at the moment. The skin is pale, with a cyanotic tint. Auscultation: vesicular breathing in the lungs, no wheezing, respiratory rate = 20 per minute. Heart: clear tones, regular rhythm, systolic murmur in the projection of the aortic valve. BP=110/70 mm Hg. art., heart rate = 78 beats/min. The abdomen is soft and painless. The liver is +3.0 cm from under the costal arch, the edge is dense, the spleen is not enlarged. Oxygen saturation - 84–87%. 6-minute walk test - 174 m. The patient is constantly taking torasemide 5 mg/day, eplerenone 25 mg/day, diltiazem 90 mg/day, warfarin 2.5 mg/day under the control of an INR of 2.0–3.0.
General blood test: hemoglobin - 161 g/l, erythrocytes - 7.6 × 1012/l, hematocrit - 56%, average erythrocyte volume - 73.6 fl, average hemoglobin content in an erythrocyte - 21.1 pg, average hemoglobin concentration in erythrocytes - 287 g/l, leukocytes - 4.8x109/l, platelets - 160x109/l.
Biochemical blood test: total protein - 75 g/l, creatinine - 58 µmol/l, uric acid - 394.8 µmol/l, aspartate aminotransferase (AST) - 16.1 U/l, alanine aminotransferase (ALT) - 15.6 U /l, total bilirubin - 10.5 µmol/l, potassium - 4.3 mmol/l.
EchoCG: no violations of local contractility. Left ventricular ejection fraction - 65%. Dilatation of the left and right atrium, right ventricle, multiple ventricular septal defects, bicuspid aortic valve. Eisenmenger syndrome. High pulmonary hypertension (average pulmonary artery pressure - 88 mm Hg). Moderate aortic valve stenosis.
Taking into account the lack of compensation and the ineffectiveness of the therapy, the patient was prescribed Bozenex 62.5 mg twice a day under the control of AST, ALT, and bilirubin. According to the recommendations, bosentan can be recommended for patients with Eisenmenger syndrome FC 3 according to WHO (class and level of evidence 1B) [12].
After 8 weeks the patient did not report any adverse events. 6-minute walk test - increase in distance by 89 m. EchoCG - no dynamics, average pressure in the pulmonary artery - 80 mm Hg. Art. All attempts to increase the dose of Bozenex were accompanied by intense headaches; the patient continued taking the drug at a dose of 62.5 mg 2 times a day.
After 12 months the patient did not report any adverse events. 6-minute walk test - increase in distance compared to the initial visit by 97 m. EchoCG - no dynamics, average pressure in the pulmonary artery - 83 mm Hg. Art. The rest of the therapy remains unchanged.
Classification
Left ventricular failure
- Arterial hypertension
- Myocarditis
- Cardiomyopathies
- Mitral regurgitation
- Coarctation of the aorta, aortic valve defects
- Cardiac ischemia
Increased pressure in the left atrium
- Tumor or thrombosis of the left atrium
- Mitral stenosis
- Supravalvular mitral annulus, triatrial heart
Pulmonary vein obstruction
- Pulmonary venous thrombosis
- Mediastinal fibrosis
Parenchymal lung diseases
- Interstitial lung diseases
- Chronic obstructive pulmonary diseases (COPD)
- Acute severe lung injury (severe diffuse pneumonitis, adult respiratory distress syndrome)
Diseases of the pulmonary artery system
- Repeated or massive pulmonary embolisms
- Primary pulmonary hypertension
- Systemic vasculitis
- Thrombosis "in situ" of the pulmonary artery
- Increased pulmonary blood flow volume (patent ductus arteriosus, congenital heart disease with left-to-right shunting)
- Distal pulmonary stenosis
- Pulmonary hypertension caused by food or drugs
Pulmonary hypertension of newborns
- Hyaline membrane disease
- Persistent fetal circulation
- Meconium aspiration
- Diaphragmatic hernia
Hypoxia and/or hypercapnia
- Obstruction of the upper respiratory tract (obstructive sleep apnea syndrome, enlarged tonsils)
- Accommodation high in the mountains
- Primary alveolar hypoventilation
- Pickwick's syndrome (hypoventilation in fat people)
Many authors classify such forms of pulmonary hypertension as acute and chronic based on the timing of development.